Pre-Market Cybersecurity Labeling: Ultimate Guide
Post Summary
Medical devices connected to networks introduce cybersecurity risks. Pre-market cybersecurity labeling, now required by the FDA, ensures these devices are secure before deployment. Here's what you need to know:
- What it is: Documentation detailing cybersecurity measures for medical devices with internet or software connectivity.
- Why it matters: Helps healthcare organizations (HDOs) manage risks, prevent cyberattacks, and ensure patient safety.
- Key requirements: Includes a Software Bill of Materials (SBOM), system architecture details, and security protocols.
- Regulatory updates: FDA mandates labeling under Section 524B of the FD&C Act, with strict guidelines since October 2023.
- Compliance risks: Missing or incomplete labeling can lead to rejected FDA submissions or device recalls.
This guide breaks down the essentials, regulatory frameworks, and best practices for creating effective cybersecurity labeling.
Cybersecurity for Medical Devices – FDA 510(k) Cyberecurity Compliance | In-Focus

sbb-itb-535baee
Key Regulatory and Standards Frameworks
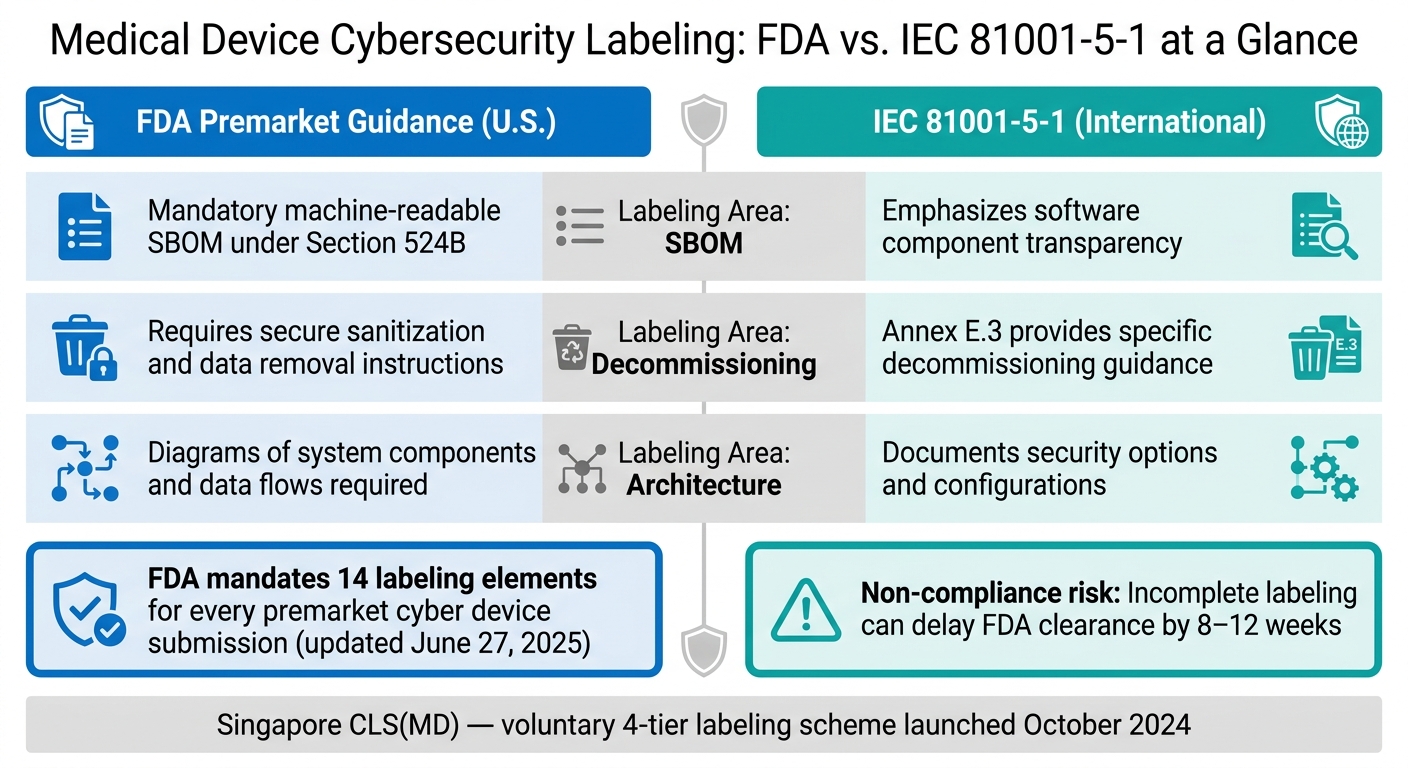
FDA vs. IEC 81001-5-1: Medical Device Cybersecurity Labeling Requirements Compared
FDA Cybersecurity Labeling Requirements
The FDA updated its premarket cybersecurity guidance on June 27, 2025, requiring manufacturers to include 14 detailed labeling elements in every premarket submission for cyber devices [4]. These elements are non-negotiable and cover a range of critical details, such as system architecture diagrams, network port inventories, forensic logging capabilities, and End-of-Support (EOS) timelines.
"Cybersecurity labeling connects design intent to real-world device use. It communicates to clinicians, IT administrators, and healthcare delivery organizations how to install, configure, maintain, and retire a device securely." - Garrett Schumacher, MS, CISSP, Velentium Medical [3]
Manufacturers must also address the device's compatibility with enterprise SIEM systems and provide clear EOS dates [3]. A practical tip? Pinpoint which of the 14 elements require user configuration or awareness during the design phase. Rushing to address labeling late in the process often leads to vague or inconsistent information, which can complicate FDA reviews [3].
In addition to U.S. regulations, international standards also play a role in shaping cybersecurity labeling practices.
International Standards for Cybersecurity Labeling
While the FDA's guidance governs U.S. submissions, international frameworks like IEC 81001-5-1 and voluntary initiatives, such as Singapore's Cybersecurity Labelling Scheme, help establish global best practices. These standards are valuable to understand, even for those focused on domestic compliance.
IEC 81001-5-1:2021 is an FDA-recognized standard that aligns closely with U.S. requirements. Its Clause 5.8 and Annex E focus on secure operation documentation, configuration guidance, and decommissioning procedures - paralleling many of the FDA's 14 labeling elements [3]. Here's how the two frameworks compare:
| Labeling Area | FDA Premarket Guidance (U.S.) | IEC 81001-5-1 (International) |
|---|---|---|
| SBOM | Mandatory machine-readable SBOM under Section 524B | Emphasizes software component transparency |
| Decommissioning | Requires secure sanitization and data removal instructions | Annex E.3 provides specific decommissioning guidance |
| Architecture | Diagrams of system components and data flows required | Documents security options and configurations |
The NEMA MDS2 form (ANSI/NEMA HN 1-2019) is another useful tool. This standardized disclosure format provides a consistent summary of a device's security controls and is often expected during procurement, even though it’s optional [3]. Singapore has also introduced the Cybersecurity Labelling Scheme for Medical Devices (CLS[MD]), launched in October 2024. This voluntary program evaluates devices across four security tiers, showing that global labeling expectations are becoming more defined [5].
Balancing Transparency and Security in Labeling
When applying regulatory and international standards, manufacturers must carefully balance transparency with security. Share too little, and healthcare delivery organizations (HDOs) can't manage risks effectively. Share too much, and you risk giving attackers a playbook. The key is to practice selective transparency.
The Vulnerability Exploitability eXchange (VEX) framework offers a practical approach to this challenge. VEX allows manufacturers to classify known vulnerabilities as "Not Affected", "Affected", "Fixed", or "Under Investigation." This lets HDOs identify a CVE in an SBOM component while understanding its limited threat due to specific design constraints [6]. It’s actionable information without overexposing sensitive details.
For risks that can't be fully eliminated, manufacturers should document compensating controls - like network segmentation, authentication protocols, or physical access restrictions - instead of diving into exploit mechanics [6]. According to Medcrypt's 2026 analysis, the FDA frequently flags submissions with "unresolved findings" that lack clear mitigation strategies or rationale [6]. The message is clear: the FDA expects manufacturers to not only identify risks but also demonstrate thoughtful responses. Aligning regulatory and international frameworks can help achieve that balance effectively.
Core Components of Cybersecurity Labeling
Device Security Architecture and Network Integration
The documentation of a device's security architecture forms the backbone of a compliant cybersecurity label. Manufacturers must include clear diagrams that show how the device operates within a healthcare network. This isn’t just about meeting regulations - it provides IT teams with the critical details they need to safely deploy and manage the device.
Transparency about interfaces is just as important. Every physical and logical port - whether it’s USB, Ethernet, Bluetooth, or serial - should be listed, along with its purpose and default status. Even interfaces that are disabled by default must be disclosed, giving healthcare delivery organizations (HDOs) a full understanding of the device’s potential vulnerabilities [3]. Diagrams showing the relationships between various components, such as implants, programmers, and cloud gateways, can help IT administrators quickly grasp deployment needs.
The labeling should also clearly outline which security responsibilities belong to the manufacturer and which fall to the HDO. For example, the manufacturer might secure device firmware, while the HDO might need to configure firewall settings or integrate with Active Directory. Any ambiguity in these roles can lead to operational risks [3]. This clarity also lays the groundwork for effective software lifecycle management.
Software Lifecycle and Update Transparency
A secure architecture is only part of the equation - software management also needs to be clearly documented. According to FDA requirements, labeling must explain how updates are delivered, authenticated, and communicated when vulnerabilities are identified [3]. As of October 1, 2023, these requirements apply to all cyber device submissions [1].
The Software Bill of Materials (SBOM) plays a central role here. Under Section 524B of the FD&C Act, manufacturers must provide a machine-readable SBOM, as outlined in earlier sections [1]. By hosting the SBOM on a manufacturer portal and updating it with each software release, HDOs can always access the latest version for their devices. Additionally, manufacturers must communicate End-of-Support (EOS) timelines upfront, allowing HDOs to plan for the device’s eventual retirement [3].
"Labeling that does not include sufficient information to explain how to securely configure or update the device may limit the ability of end users to appropriately manage and protect the medical device system." - FDA [7]
Comprehensive software lifecycle documentation enables HDOs to securely manage devices from initial deployment to end-of-life.
Security Features and Residual Risk Communication
Security features should not just be listed - they need to be explained. Labeling should detail how these features protect patients and clinical workflows during a cyberattack. For example, manufacturers should describe the controls designed to maintain essential performance during an attack and provide clear configuration instructions for authentication methods, encryption standards, and secure default settings [2][3].
This kind of transparency not only meets regulatory requirements but also gives HDOs actionable information. Communicating residual risks is equally important. A 2026 report revealed that 24% of healthcare organizations experienced cyberattacks targeting medical devices, with 80% of these incidents affecting patient care [6]. Such statistics underscore the importance of labeling that clearly explains residual risks, especially those introduced by user actions - like connecting to an unapproved network. Manufacturers should also provide indicators of compromise, such as unexpected device restarts or communication errors, to help clinical staff identify potential security breaches [6].
The table below outlines the key components of cybersecurity labeling along with the essential details they should include:
| Labeling Component | Essential Elements to Include |
|---|---|
| Network Interfaces | List all physical/logical ports, their purpose, and default status (enabled/disabled) [3] |
| Update Process | Include delivery mechanism, authentication method, version control, and notification steps [3] |
| Forensic Logging | Specify log formats (e.g., CEF, JSON, syslog), data retention policies, and SIEM compatibility [3] |
| Decommissioning | Provide step-by-step instructions for data sanitization, key revocation, and physical destruction [3] |
| SBOM | Detail commercial, open-source, and off-the-shelf software components [1][3] |
Manufacturers are advised to ship devices in their most secure default configuration and to include explanations for these settings in the labeling. When HDOs understand the rationale behind a default, they are less likely to alter it in ways that could create new vulnerabilities.
Best Practices for Building and Maintaining Cybersecurity Labeling
Structuring Labeling for Different Stakeholders
Cybersecurity labeling needs to cater to a variety of audiences, including clinicians, IT administrators, biomedical engineers, and procurement staff. Each group has distinct needs:
- Clinicians require straightforward instructions to identify anomalies and ensure device safety.
- IT administrators need technical details like port lists, network diagrams, and compatibility with security tools like SIEM.
- Biomedical engineers focus on maintenance procedures and secure methods for decommissioning devices.
- Procurement staff rely on documents such as the SBOM, MDS2 forms, and End-of-Support timelines to make informed buying decisions.
As Velentium Medical explains:
"Cybersecurity labeling connects design intent to real-world device use. It communicates to clinicians, IT administrators, and healthcare delivery organizations how to install, configure, maintain, and retire a device securely." - Velentium Medical [3]
To meet these diverse needs, organize labeling into sections specific to each audience. For clinical staff, use clear, action-oriented instructions like "Only connect the device to approved hospital networks" or "Report unexplained restarts to the manufacturer immediately." On the other hand, IT-focused documentation should include technical diagrams and detailed tables, helping IT teams quickly find the information they need without confusion. Including standardized forms, such as the MDS2, ensures smoother communication across teams.
When labeling is tailored to its audience, it not only improves usability but also supports effective maintenance and security updates.
Lifecycle Maintenance and Change Control
Effective cybersecurity labeling doesn’t end at product launch - it’s a continuous responsibility throughout the device's lifecycle.
As Wayne Stewart, Vice President, Global – IoT & AI at Intertek, explains:
"Cybersecurity is now evaluated as a lifecycle obligation rather than a static design feature assessed only at the point of market entry." [8]
This means every update - whether it’s a routine feature enhancement or a critical security patch - should trigger a review of the labeling. The table below outlines how different updates impact labeling requirements:
| Update Type | Required Labeling Action |
|---|---|
| Routine Feature Update | Update version numbers; ensure Instructions for Use (IFU) align. |
| Security Patch | Revise the "Authorized Update Process" section; notify users. |
| New Vulnerability (Affected) | Update the Unresolved Anomalies table; add compensating controls to the IFU. |
| Architecture Change | Update system diagrams and revise the MDS2 form. |
Traceability is key. For example, unresolved vulnerabilities should link to specific SBOM components using unique identifiers like package URLs. FDA reviewers often scrutinize unresolved issues, even those deemed low or medium risk, if they lack documented mitigations or clinical impact analyses.
As Ran Chen, Global MedTech Expert, points out:
"A well-documented mitigation strategy is more credible than an empty table on a 500,000-line codebase."
When adding compensating controls, be precise. A statement like "Diagnostic port restricted to service-mode only" is far more actionable than a vague mention of "network segmentation."
Using Platforms to Support Cyber Risk Management
Once labeling structure and update processes are in place, integrating this information into a centralized risk management platform can simplify managing device security.
Even with well-organized labeling, keeping track of critical medical device security risks like SBOMs, End-of-Support dates, and unresolved vulnerabilities across multiple devices is complex. This is where platforms like Censinet RiskOps™ come in.
Censinet RiskOps™ helps healthcare organizations streamline risk assessments, evaluate the cybersecurity posture of devices and vendors, and collaborate across clinical, IT, and procurement teams. By centralizing device risk data - including information from manufacturer labeling - organizations can shift from reactive responses to proactive risk management.
The platform’s Censinet AITM feature further simplifies the process by automating vendor security questionnaires and summarizing relevant documentation. This approach transforms cybersecurity labeling into a practical tool for managing risks effectively.
Conclusion
Pre-market cybersecurity labeling has transitioned from being a mere compliance checkbox to a critical safety measure. Without adequate cybersecurity information, devices risk being misbranded under Section 502(f), potentially leading to market withdrawal. For manufacturers and healthcare delivery organizations (HDOs), this poses serious operational, financial, and patient safety risks. On top of that, gaps in cybersecurity documentation during premarket submissions - such as incomplete labeling - can delay clearance timelines by 8 to 12 weeks [2]. This makes thorough and accurate documentation a pressing business priority. This is especially true when conducting third-party vendor risk management for new medical devices.
This shift in labeling practices also impacts how clinical teams and IT administrators approach device security. By including elements like clear instructions for accessing Software Bill of Materials (SBOM), end-of-support timelines, and hardening guidelines, manufacturers equip HDOs to address vulnerabilities proactively instead of waiting for incidents to occur.
"Cybersecurity threats to the healthcare sector have become more frequent and more severe, carrying increased potential for clinical impact." - FDA [7]
The industry is moving toward treating labeling as an ongoing responsibility. This means embedding it into quality management systems and updating it with every patch or architecture change. Tools like machine-readable SBOMs, Vulnerability Exploitability eXchange (VEX) documents, and structured vulnerability disclosure policies are becoming standard practices. This lifecycle-focused approach ensures devices maintain a strong security posture as they evolve, meeting both regulatory and operational expectations.
Platforms like Censinet RiskOps™ take this a step further by turning labeling data into actionable insights. By centralizing device information, automating risk assessments, and enabling proactive decisions across clinical, IT, and procurement teams, these platforms enhance the effectiveness of cybersecurity labeling. Together, clear labeling and tools like Censinet RiskOps™ strengthen the healthcare ecosystem, making it safer and more resilient.
FAQs
Does my device qualify as an FDA “cyber device” under Section 524B?
Your device falls under the FDA's definition of a "cyber device" as outlined in Section 524B if it meets the following criteria: it includes software, connects to the internet or other networks, and has features that could be exposed to cybersecurity threats. These guidelines are in place to ensure devices are evaluated for potential risks and adhere to cybersecurity requirements.
What should my machine-readable SBOM and VEX include for FDA review?
When submitting an SBOM (Software Bill of Materials) for FDA review, it must comprehensively list all software components in the medical device. This includes:
- Proprietary software
- Open-source modules
- Third-party components
To meet FDA standards, your SBOM should adhere to formats like SPDX or CycloneDX. It must also provide key details for each component, such as:
- Version: The specific version of the software.
- Origin: Where the software comes from.
- Support level: The type of support available for the software.
- End-of-support date: When support for the software will cease.
Instead of embedding the SBOM directly into device labels, include a summary of third-party components and provide clear instructions for accessing the full SBOM.
Incorporating VEX Files for Vulnerability Management
To address potential vulnerabilities, include Vulnerability Exploitability eXchange (VEX) files. These files clarify whether identified vulnerabilities (CVEs) are exploitable within the device's specific configuration.
Key points for VEX documentation:
- Keep VEX files updated as new vulnerabilities are identified.
- Ensure both SBOM and VEX files are machine-readable to support automated risk management processes and align with FDA transparency expectations.
By following these guidelines, you'll ensure your submission meets the FDA's requirements for software transparency and risk management.
How do I keep cybersecurity labeling current after patches, vulnerabilities, or architecture changes?
To keep cybersecurity labeling current after patches, vulnerabilities, or system changes, manufacturers need to follow FDA guidance and update the labeling within 30 days of identifying a security issue. Here's how they can handle this process effectively:
- Record updates: Document newly identified CVEs, patches, or configuration adjustments.
- Disclose vulnerabilities: Share details about any unresolved vulnerabilities, along with compensating controls in place to mitigate risks.
- Review regularly: Conduct ongoing security assessments and ensure any major changes are promptly reflected in the labeling.
These steps help maintain compliance while supporting effective lifecycle cybersecurity management.

