The FDA has issued updated guidelines to strengthen cybersecurity for internet-connected medical devices. These rules, effective February 2, 2026, require manufacturers to actively monitor, disclose, and address vulnerabilities throughout a device's lifecycle. Key requirements include:
These measures align with the FDA's updated Quality Management System Regulation (QMSR), integrating cybersecurity into risk management, design, and post-market surveillance. Both manufacturers and healthcare organizations must collaborate to safeguard devices and patient safety. The goal: prevent cyber threats from disrupting care.
A Quick Primer on FDA's Final Guidance for Cybersecurity in Medical Devices
sbb-itb-535baee
FDA Post-Market Cybersecurity Requirements
Since March 29, 2023, the FDA's post-market cybersecurity framework, under Section 524B of the FD&C Act, has set specific legal requirements for managing the lifecycle of "cyber devices." These devices include software, internet connectivity (such as USB or Bluetooth), and other technological features that make them susceptible to cyber threats [3].
To comply, manufacturers must submit comprehensive cybersecurity management plans to the FDA. These plans must outline how they will monitor vulnerabilities, implement coordinated disclosure processes, and provide timely security updates. Without these plans, devices meeting the cyber device definition cannot gain market authorization [3]. Below, we break down the three key obligations manufacturers must meet.
Continuous Vulnerability Monitoring
Manufacturers are required to actively monitor for cybersecurity threats. This involves tracking internal systems, consulting CERTs (like ICS-CERT), joining ISAOs, and reviewing reports from independent researchers [1]. The FDA recommends joining ISAOs such as the Health Information Sharing & Analysis Center (H-ISAC) for access to up-to-the-minute threat intelligence [1].
A machine-readable Software Bill of Materials (SBOM), aligned with the 2021 NTIA "minimum elements" standard, is essential. This document lists software components and their support timelines, helping manufacturers quickly identify and address vulnerabilities in third-party software [3].
When vulnerabilities arise, manufacturers must evaluate their severity using a risk-based approach. This includes assessing the likelihood of exploitation, potential impact on device functionality, and enterprise risks to patient safety. Risks are classified as either "Controlled" (acceptable) or "Uncontrolled" (requiring immediate action) [1].
Coordinated Vulnerability Disclosure (CVD)
The FDA also requires manufacturers to establish a formal process for stakeholders - such as security researchers, healthcare providers, and software vendors - to report vulnerabilities without fear of legal repercussions [3]. According to Section 524B of the FD&C Act:
A person who submits an application... for a device that meets the definition of a cyber device... shall include... a plan to monitor, identify, and address, as appropriate, in a reasonable time, postmarket cybersecurity vulnerabilities and exploits, including coordinated vulnerability disclosure and related procedures
.
These processes should include clear channels for receiving and evaluating vulnerability reports, often using tools like the Common Vulnerability Scoring System (CVSS) [4]. Manufacturers are also expected to communicate both vulnerabilities and mitigations to users, patients, and relevant federal agencies. Importantly, proactive cybersecurity updates made under a CVD program typically do not need to be reported to the FDA under 21 CFR part 806.10, provided no harm to patients occurs [4].
Patch Management and Security Updates
The FDA distinguishes between routine and critical security updates. Routine patches for known vulnerabilities should be deployed on a regular schedule [3]. However, for critical vulnerabilities that pose "uncontrolled" risks to patient safety, manufacturers must release patches as quickly as possible [3]. Most routine updates are considered device improvements and do not require FDA reporting under 21 CFR part 806 - unless they address an uncontrolled safety risk [1].
Cybersecurity in Quality Management Systems
On February 2, 2026, the FDA introduced its updated Quality Management System Regulation (QMSR), replacing the older Quality System Regulation (QSR) under 21 CFR Part 820. This new regulation incorporates ISO 13485:2016 by reference and weaves cybersecurity into key areas like risk management, design controls, validation, and post-market surveillance [7]. Just a day later, on February 3, 2026, the FDA revised its cybersecurity guidance to align with these fresh requirements [8].
Under this updated framework, cybersecurity is no longer just a technical afterthought. It is now a mandatory element of quality systems, directly tied to patient safety and device performance. Known vulnerabilities are classified as "reasonably foreseeable risks", requiring proactive management within the quality system.
"Cybersecurity is no longer a standalone technical consideration - it is embedded into Risk Management, Design Controls, Validation Activities, and Post-market Surveillance."
–
This regulatory overhaul also changes how the FDA conducts inspections. The old QSIT-based framework has been replaced with a risk-based QMS audit approach (Compliance Program 7382.850), which mirrors the MDSAP audit methodology. This new style focuses on a system-level review, diving deep into risk files, management reviews, and trends identified in post-market surveillance. The emphasis on cybersecurity throughout the product lifecycle underscores the importance of continuous vigilance. For manufacturers, this means adopting a strong documentation strategy to prove compliance under these updated standards.
Documentation and Compliance Requirements
Meeting these integrated cybersecurity obligations requires manufacturers to revamp their documentation practices. Detailed records demonstrating QMSR compliance are now essential. These include:
An accurate SBOM is especially critical - any gaps in this documentation could violate Section 301(q) of the FD&C Act.
Additionally, manufacturers must show how cybersecurity is embedded across various ISO 13485 clauses. For instance, Clause 7.3.7 mandates that design validation include security testing, while Clause 8.5 requires integrating corrective and preventive action (CAPA) processes with security events and identified vulnerabilities. Quality Manuals, SOPs, and Design History Files must be updated to reflect cybersecurity as a core design input and validation requirement throughout the Total Product Lifecycle.
Manufacturer and Healthcare Organization Collaboration
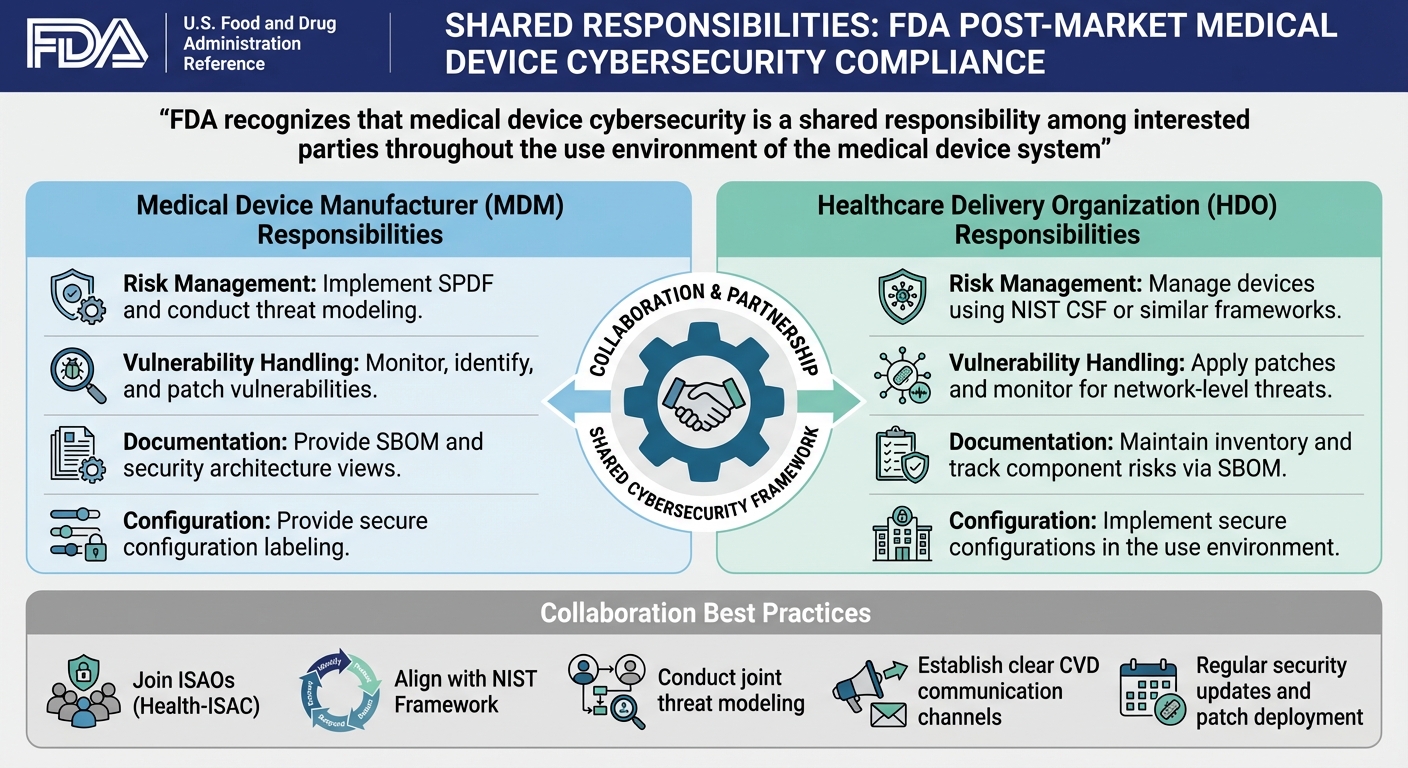
FDA Medical Device Cybersecurity: Manufacturer vs Healthcare Organization Responsibilities
The FDA stresses that medical device cybersecurity is a team effort. It involves collaboration among medical device manufacturers (MDMs), healthcare delivery organizations (HDOs), patients, and healthcare providers throughout the device's lifecycle [2].
"FDA recognizes that
Real-world cases have highlighted the importance of this shared responsibility. Both MDMs and HDOs need to approach the medical device system as a connected ecosystem. This includes the device itself, the facility's network, and the supporting infrastructure. Treating the device as a standalone element simply doesn’t work in today’s interconnected healthcare environments [2]. Meeting the FDA's updated cybersecurity requirements depends on this coordinated effort. Let’s break down the distinct roles both manufacturers and healthcare organizations play.
Roles and Responsibilities
MDMs and HDOs each have specific responsibilities that complement one another in maintaining cybersecurity after a device hits the market.
MDMs are tasked with designing and maintaining devices using a Secure Product Development Framework (SPDF). This framework addresses vulnerabilities throughout the device’s lifecycle - from initial design to eventual decommissioning. Transparency is crucial here. MDMs need to provide clear documentation, such as a Software Bill of Materials (SBOM), and disclose information about communication interfaces and third-party software components. Devices must meet key security goals, including authenticity, authorization, availability, confidentiality, and the ability to receive secure and timely updates [2].
On the other hand, HDOs focus on securely integrating medical devices into their environments - like hospital networks - and managing third-party risk and cybersecurity throughout the device's lifespan. This involves applying updates and patches provided by manufacturers, adhering to frameworks like the NIST Framework for Improving Critical Infrastructure Cybersecurity, and ensuring devices are configured, installed, and monitored securely. Regular log reviews also fall under their purview [2]. Together, these roles align to meet the FDA’s requirements for monitoring vulnerabilities and managing patches.
Responsibility Area
Manufacturer (MDM)
Healthcare Organization (HDO)
Implement SPDF and conduct threat modeling
Manage devices using NIST CSF or similar frameworks
Monitor, identify, and patch vulnerabilities
Apply patches and monitor for network-level threats
Provide SBOM and security architecture views
Maintain inventory and track component risks via SBOM
Provide secure configuration labeling
Implement secure configurations in the use environment
One critical point to note: inadequate labeling can lead to compliance issues. If a manufacturer fails to provide sufficient instructions for secure configuration or updates, the FDA may classify the device as misbranded [2]. With clear roles established, collaboration is the next step in strengthening cybersecurity.
Collaboration Best Practices
Effective collaboration goes beyond fulfilling individual responsibilities. It requires shared, practical strategies. The FDA encourages voluntary participation in Information Sharing Analysis Organizations (ISAOs), such as the Health Information Sharing & Analysis Center (Health-ISAC). These groups play a key role in building a strong post-market cybersecurity program [1]. Aligning efforts with frameworks like the NIST Framework (Identify, Protect, Detect, Respond, and Recover) helps both MDMs and HDOs speak the same language when managing cybersecurity. It also ensures that security updates and patches are regularly provided by MDMs and swiftly applied by HDOs [1].
Joint threat modeling is another essential practice. By working together to identify vulnerabilities and establish countermeasures, MDMs and HDOs can reduce risks and prevent harm to patients. When built-in device controls fall short, compensating measures - like enhanced network configurations or isolation strategies - become critical [1]. Lastly, having clear communication channels for Coordinated Vulnerability Disclosure (CVD) ensures that any vulnerabilities discovered after a device is released are addressed in a timely and organized way [9].
How Censinet Supports Post-Market Medical Device Cybersecurity
Complying with the FDA's post-market cybersecurity requirements demands the right mix of tools and strategies. Healthcare organizations need systems that enable continuous monitoring, smooth vendor collaboration, and thorough documentation, all of which are emphasized in the FDA guidance. As stated by the FDA:
The health care environment is complex, and manufacturers, hospitals, and facilities must work together to manage cybersecurity risks
.
Specialized platforms are crucial for simplifying the process of risk assessments and fostering collaboration.
Automated Risk Assessments with Censinet RiskOps™
Censinet RiskOps™ tackles one of the biggest hurdles in FDA compliance: conducting risk assessments efficiently. It automates both third-party and enterprise risk assessments, giving healthcare delivery organizations the ability to evaluate medical device vendors and their products against cybersecurity standards without relying on time-consuming manual processes.
With Censinet AITM™, vendors can complete security questionnaires quickly, while the platform automatically summarizes evidence and documentation. It also captures essential details about product integration and fourth-party risks. For organizations managing numerous medical devices, this automation significantly boosts efficiency and compliance. Importantly, configurable rules and review processes ensure that automation complements human judgment rather than replacing it.
Collaboration and Risk Management Tools
Cybersecurity is a shared responsibility between manufacturers and healthcare organizations, and Censinet RiskOps™ facilitates this partnership through Censinet Connect™. This tool provides a direct communication channel for healthcare organizations and medical device vendors, streamlining coordinated risk management efforts. It supports activities like vulnerability disclosure (CVD) and patch management, which are vital for maintaining cybersecurity.
The platform's command center offers real-time risk visualization, enabling both parties to share actionable insights and prioritize vulnerabilities together. By centralizing information on device risks, policies, and tasks, Censinet RiskOps™ serves as a central hub for managing post-market device cybersecurity effectively.
Conclusion
The FDA's post-market cybersecurity guidance leaves no room for doubt: cybersecurity is a core element of medical device safety. As the FDA puts it, "Ensuring device safety and effectiveness includes adequate device cybersecurity, as well as its security as part of the larger system" [2]. Real-world incidents have shown how vulnerabilities in connected systems can jeopardize patient care, underscoring the direct link between cyber risks and patient safety.
This framework goes beyond regulatory compliance - it’s about protecting lives. While adherence to the Quality Management System Regulation (QMSR) and Section 524B of the FD&C Act is required, the ultimate goal is to prevent harm to patients. The FDA focuses on addressing "uncontrolled risks", where the likelihood and severity of potential harm are simply unacceptable [10]. In today’s interconnected healthcare environment, where medical devices are part of broader hospital networks, a single vulnerability can ripple through systems, disrupting care on a large scale.
To protect these systems, shared responsibility is key. Continuous monitoring, coordinated vulnerability disclosure, and rapid patching are critical steps. Joining an Information Sharing Analysis Organization (ISAO) allows organizations to stay informed about evolving threats. Additionally, addressing vulnerabilities within the FDA’s recommended timelines - communicating issues within 30 days and resolving them within 60 days - can help avoid certain enforcement actions [10].
Cybersecurity must be embedded at every stage of device management. Vulnerabilities like URGENT/11 and SweynTooth, which affect third-party software across various devices and clinical areas [2], highlight the importance of proactive risk management across the entire product lifecycle. Meeting FDA standards not only ensures compliance but also strengthens operational resilience, ultimately safeguarding patients.
FAQs
Does my device qualify as a “cyber device” under Section 524B?
Devices are classified as “cyber devices” under Section 524B if they feature internet connectivity, updatable software, or rely on systems like update servers. These devices must also address cybersecurity in their premarket submissions and lifecycle management. To determine if your device falls into this category, consult the FDA's expanded definition for clarification.
What evidence does the FDA expect during QMSR audits for cybersecurity?
The FDA requires documented evidence of cybersecurity measures for medical devices. This includes conducting risk management assessments to evaluate both exploitability and potential impact. Additionally, manufacturers must show they have processes in place for ongoing vulnerability monitoring. These steps are essential to meet the post-market cybersecurity compliance standards set by the FDA.
How quickly must we disclose and address 'uncontrolled' vulnerabilities?
According to the FDA's 2016 guidance, medical device manufacturers are expected to address uncontrolled vulnerabilities within 60 days. Additionally, customers must be informed within 30 days to ensure prompt communication and effective risk management.
Related Blog Posts
- IoT Device Compliance: FDA Postmarket Audit Guide
- Best Practices for FDA IoT Cybersecurity Compliance
- 5 Key Premarket Cybersecurity Requirements for Devices
- How FDA Cybersecurity Guidance Impacts HDOs
{"@context":"https://schema.org","@type":"FAQPage","mainEntity":[{"@type":"Question","name":"Does my device qualify as a “cyber device” under Section 524B?","acceptedAnswer":{"@type":"Answer","text":"<p>Devices are classified as “cyber devices” under Section 524B if they feature <strong>internet connectivity</strong>, <strong>updatable software</strong>, or rely on systems like update servers. These devices must also address <strong>cybersecurity</strong> in their premarket submissions and lifecycle management. To determine if your device falls into this category, consult the FDA's expanded definition for clarification.</p>"}},{"@type":"Question","name":"What evidence does the FDA expect during QMSR audits for cybersecurity?","acceptedAnswer":{"@type":"Answer","text":"<p>The FDA requires <strong>documented evidence</strong> of cybersecurity measures for medical devices. This includes conducting <strong>risk management assessments</strong> to evaluate both exploitability and potential impact. Additionally, manufacturers must show they have processes in place for <strong>ongoing vulnerability monitoring</strong>. These steps are essential to meet the post-market cybersecurity compliance standards set by the FDA.</p>"}},{"@type":"Question","name":"How quickly must we disclose and address 'uncontrolled' vulnerabilities?","acceptedAnswer":{"@type":"Answer","text":"<p>According to the FDA's 2016 guidance, medical device manufacturers are expected to address <em>uncontrolled</em> vulnerabilities within <strong>60 days</strong>. Additionally, customers must be informed within <strong>30 days</strong> to ensure prompt communication and effective risk management.</p>"}}]}
