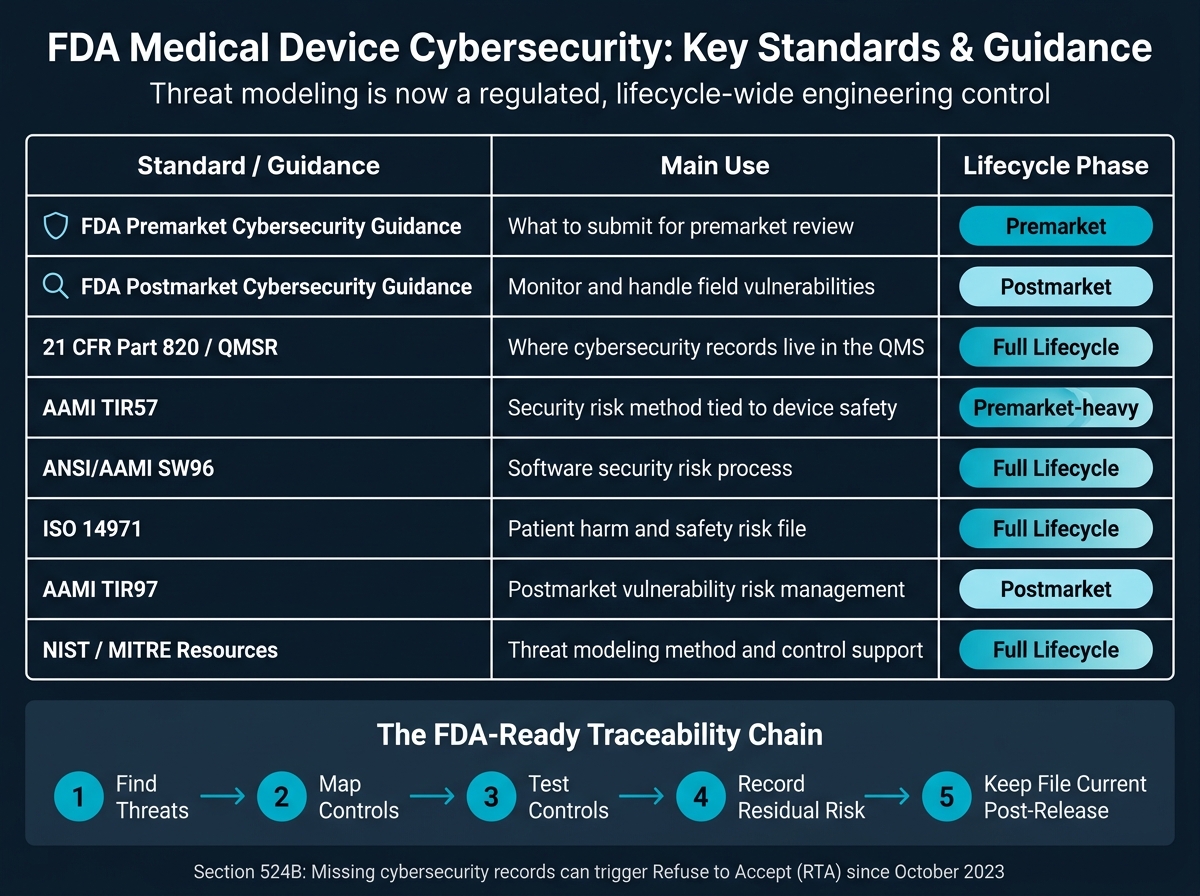
If your FDA submission does not include a threat model, it can get stopped before review even starts. Since October 2023, FDA has had Refuse to Accept authority under Section 524B for missing cybersecurity records.
If I had to boil this article down to the main point, it would be this: threat modeling is now part of medical device design control, risk files, testing, and postmarket updates. It is not just a security document. It has to connect to your QMS, your SBOM, your verification evidence, and your patient harm analysis.
Here’s what you need to know up front:
- FDA expects system-level scope, not just the device by itself
- Premarket and postmarket work connect, using the same threat model record
- Section 524B ties cybersecurity records to submission readiness
- 21 CFR Part 820 / QMSR ties threat modeling to controlled quality records
- ISO 14971 covers patient harm and residual safety risk
- AAMI TIR57 and SW96 guide security risk analysis and control mapping
- AAMI TIR97 supports postmarket vulnerability handling
- NIST and MITRE help teams build a repeatable process
A simple way to think about it: find threats, map controls, test those controls, record what risk remains, and keep the file current after release. If any link in that chain is weak, FDA may ask questions - or stop the file at intake.
Quick comparison
| Standard or guidance | Main use | Where it fits |
|---|---|---|
| FDA Premarket Cybersecurity Guidance | What to submit | Premarket |
| FDA Postmarket Guidance | How to watch and handle field issues | Postmarket |
| 21 CFR Part 820 / QMSR | Where records live in the quality system | Full lifecycle |
| AAMI TIR57 | Security risk method tied to device safety | Premarket-heavy |
| ANSI/AAMI SW96 | Software security risk process | Full lifecycle |
| ISO 14971 | Patient harm and safety risk file | Full lifecycle |
| AAMI TIR97 | Postmarket vulnerability risk work | Postmarket |
| NIST / MITRE resources | Threat modeling method and control support | Full lifecycle |
In short: a strong FDA-ready program shows traceability from threat to requirement to control to test to residual risk, then keeps that record under change control as the device and threat landscape shift.
FDA Medical Device Cybersecurity Standards: Quick Reference Guide
Threat Modeling and Risk Assessment Webinar
sbb-itb-535baee
Why Threat Modeling Matters for FDA Cybersecurity Compliance

Section 524B of the FD&C Act requires cybersecurity information in premarket submissions. That means manufacturers need to show they identified and addressed credible risks.[2]
How Threat Modeling Supports Secure Design and Risk Management
Threat modeling matters most because it starts early. When teams map out how a threat actor could compromise or degrade a device during development, they can fix weak spots in the architecture before those issues get baked in. That lines up with secure-by-design principles and helps produce the kind of documentation FDA expects in premarket submissions.[1][3]
It also helps separate security risk management from safety risk management under ISO 14971. That matters because a security control can solve one problem while creating another if safety is not considered at the same time. Threat modeling can also show how a given threat may lead to direct or indirect patient harm, including delayed diagnosis or treatment.[2]
What FDA Expects Manufacturers to Cover in Scope
FDA expects threat models to cover the full system, not just the device itself. In practice, that includes device software, hardware interfaces, cloud services, wireless and wired connections, interoperability points, and the intended use environment.[1][2]
A narrow scope leads to weak risk decisions. If assets or interfaces are left out, the model misses part of the picture. For that reason, manufacturers should document the system view, patient-impact view, update view, and security-use-case view in a single model. Third-party software and open-source components belong in scope too. Section 524B(b) also requires an SBOM, usually in SPDX or CycloneDX format.[3]
That scope definition becomes the basis for the evidence FDA expects in the submission.
Why Threat Modeling Is Part of Submission Documentation
A threat model shapes security requirements, test plans, residual risk decisions, and labeling. Those are all part of the evidence FDA expects in premarket submissions. It also serves as the baseline for postmarket updates, so the same model should carry forward into postmarket monitoring and updates.
How FDA Expects Threat Modeling to Be Used Across the Device Lifecycle
Once the premarket scope and evidence are in place, FDA does not treat threat modeling as a one-and-done exercise. It expects the same threat model to stay active through design, release, and postmarket monitoring. In plain terms, threat modeling is a continuous activity across the total product lifecycle, from early design through decommissioning. The model should be kept as a controlled, updated record across the device lifecycle.[2][3]
How Threat Modeling Is Used Before Market Submission
During design and development, threat modeling feeds straight into a Secure Product Development Framework (SPDF). That framework brings cybersecurity into the product from design through decommissioning. The key step here is traceability: modeled threats should map to security requirements, design controls, and verification and validation evidence. FDA also treats exploitation of known vulnerabilities or weak controls as a foreseeable failure mode, which means those issues need to be addressed before submission.[1][2]
FDA also expects the amount of documentation to match the level of risk. A device used in a safety-critical control loop needs far more evidence than a standalone tool. The submission package should show how the device design addresses five core security objectives:
- Authenticity (including integrity)
- Authorization
- Availability
- Confidentiality
- Updatability/Patchability[2]
Keeping Threat Models Current After Market Release
After release, the premarket threat model becomes the starting point for postmarket vulnerability management. Section 524B of the FD&C Act requires manufacturers to monitor, identify, and address postmarket vulnerabilities and exploits. So the model needs updates when vulnerabilities are disclosed in third-party components, when software is updated or patched, when device configurations change, when the intended or actual environment of use shifts, or when new attack methods appear.[3][2] FDA makes the point clearly: cybersecurity controls weaken over time as new risks and attack methods show up.
The SBOM plays a big part here. Regularly scanning it for newly disclosed vulnerabilities in open-source or third-party components is one of the most practical ways to spot when a threat model update is needed.[2]
Traceability Inside the Quality Management System
This lifecycle approach only works if the threat model is tied to the quality system. Under the QMSR effective February 2, 2026, cybersecurity documentation must stay inside controlled QMS processes.[1][3] Put simply, the work can't live in a side folder or an isolated spreadsheet. It needs to connect to the same controlled system used for the rest of the device record.
The table below shows how threat modeling artifacts map to specific ISO 13485 process requirements:
| SPDF Phase | ISO 13485 Process Mapping |
|---|---|
| Threat Modeling | Design inputs (7.3.3) |
| Secure Design & Controls | Design outputs (7.3.4) |
| Security Testing (pen testing, fuzzing) | Design verification (7.3.6) & validation (7.3.7) |
| Post-Market Monitoring | Feedback (8.2.1) & Complaint handling (8.2.2) |
| Vulnerability Response | Corrective action (8.5.2) |
Every threat, control, and test result must trace back to a controlled QMS requirement throughout the device lifecycle. This is the lifecycle approach that the standards and guidance cited in the following sections put into practice.
1. FDA Guidance: Cybersecurity in Medical Devices - Quality System Considerations and Content of Premarket Submissions
This guidance takes the lifecycle framework above and turns it into the kind of documentation FDA expects to see in a submission. Revised on February 3, 2026, it applies to 510(k), De Novo, PMA, HDE, and BLA submissions, and it links cybersecurity records directly to QMS requirements. [2]
FDA treats cybersecurity as part of device safety and effectiveness, not as something extra you tack on later.
Threat Modeling Documentation Requirements
FDA expects manufacturers to document the full system around the device, not just the device itself. That includes facility networks, update servers, nearby devices, and every outside-facing interface, such as:
- Wi-Fi
- Bluetooth
- BLE
- cellular
- USB
- Ethernet
- serial ports [2]
The guidance also says manufacturers should show how the system architecture addresses five security objectives: Authenticity (including integrity), Authorization, Availability, Confidentiality, and Secure Updatability. The depth of that evidence should line up with the device’s connectivity and the level of patient risk. [2]
Traceability to Controls, Testing, and Residual Risk
Once the scope is set, FDA wants to see a straight line from the threat model to the controls, the testing, and the remaining risk. Put simply, the agency expects clear traceability from threat to control to verification evidence. [2]
| Traceability Element | FDA Documentation Expectation |
|---|---|
| System Architecture | Visual maps of device, cloud, APIs, and third-party integrations |
| Threat Enumeration | Attack paths, actor types, exploit difficulty |
| Security Controls | Documentation of encryption, MFA, secure boot, and integrity checks |
| Verification Testing | Evidence from VAPT, fuzzing, and penetration testing focused on high-risk areas |
| Residual Risk | Why remaining risk is acceptable |
FDA does not expect zero residual risk. It expects a reasoned explanation for why the risk that remains is acceptable. [2]
2. FDA Postmarket Management of Cybersecurity in Medical Devices Guidance
FDA's 2016 Postmarket Management of Cybersecurity in Medical Devices guidance picks up where premarket work leaves off. The premarket model maps the attack paths you expect before launch. This guidance explains how manufacturers keep that model up to date once the device is out in the field.
That shift matters. Once a device is deployed, cybersecurity work stops being about lab conditions and starts being about what happens in actual use. That field view shapes how postmarket vulnerabilities are watched, sorted, and recorded.
Premarket vs. Postmarket Lifecycle Relevance
After release, threat models need to reflect field environments such as hospital networks, home networks, update servers, and connected devices that may not have been part of the original design environment. [1][2]
Under Section 524B of the FD&C Act, manufacturers of "cyber devices" must submit and maintain a plan to monitor, identify, and address postmarket vulnerabilities and exploits in a reasonable timeframe. [4] That plan must also include Coordinated Vulnerability Disclosure (CVD) procedures to handle reports of newly discovered threats.
Threat Modeling Documentation Requirements
When a new vulnerability surfaces in a third-party component listed in the device's Software Bill of Materials (SBOM), the threat model should be updated to show the changed attack surface, exploitability, and possible patient impact. [2] In practice, third-party component issues are one of the clearest signals that a postmarket model needs attention.
Traceability to Controls, Testing, and Residual Risk
Each field-identified threat should tie back to a compensating control, validation evidence such as penetration testing or vulnerability scanning, and a written reason for any residual risk that still remains. [2] Predetermined Change Control Plans (PCCPs) can help move patches into deployment faster when new field vulnerabilities appear. [1]
Those postmarket records must remain controlled in the QMS, where the next standard sets the rules for change control and corrective action.
3. 21 CFR Part 820 Quality System Regulation
Part 820 is where threat modeling moves from a planning exercise into controlled design records and postmarket proof.
Applicability
Under the QMSR in 21 CFR Part 820, cybersecurity sits inside the quality system. It is not a separate submission add-on. Under 21 CFR 820.10(c), this applies to Class II and III devices, plus Class I devices that are automated with computer software. [10] For cyber devices under Section 524B, SBOM and update-plan records also belong in the QMS. [2][8]
Threat Modeling Documentation Requirements
Under the QMSR, threat modeling is a Design Input under ISO 13485 Subclause 7.3.3. That means security requirements should come out of threat analysis during design, not after release. [8]
| Cybersecurity Activity | ISO 13485 / QMSR Clause | Record Type |
|---|---|---|
| Threat Modeling | Design Inputs (7.3.3) | Security requirements derived from analysis |
| Security Architecture | Design Outputs (7.3.4) | System diagrams and trust boundaries |
| Vulnerability Testing | Design Verification (7.3.6) | Penetration test reports, fuzzing results |
| Residual Risk Decisions | Risk Management (7.1) | Security Risk Management File |
| Patch Management | Corrective Action (8.5.2) | Post-market vulnerability response records |
Traceability to Controls, Testing, and Residual Risk
The key idea is traceability. A threat model should tie back to 7.3.3 design inputs, connect to 7.3.4 design outputs, and link forward to 7.3.6 and 7.3.7 test evidence. [8] If a team identifies a threat, there should be a clear line from that threat to the control, then to the test that shows the control works.
Residual-risk decisions need to be recorded in the security risk file and matched to ISO 14971. [2] Postmarket monitoring and patching should be recorded in CAPA under 8.5.2. [8]
Part 820 sets the record structure. ISO 14971 sets the risk process behind it.
4. AAMI TIR57: Principles for Medical Device Security - Risk Management
If Part 820 tells you where risk records live, TIR57 tells you how to think through security risk inside those records.
AAMI TIR57 is an older framework for security risk management in medical devices. It was built to connect IT security with medical device safety. Even though ANSI/AAMI SW96:2023 has now taken its place in many cases, TIR57 still matters as a reference point for understanding how security risk fits into an ISO 14971-based quality system. [11]
FDA Relevance
FDA's 2023 cybersecurity guidance explicitly references TIR57 as a resource for developing a cybersecurity-specific risk management plan and report. [12] That matters because TIR57 turns broad FDA guidance into exploitability-based security risk records.
In plain English, TIR57 adds security-focused analysis to ISO 14971. So if a manufacturer already has a quality system built around ISO 14971, TIR57 gives them a practical way to fold cybersecurity risk management into that setup without starting from scratch.
Threat Modeling Documentation Requirements
One of the biggest shifts in TIR57 is this: it treats exploitability, not probability, as the basis for cybersecurity risk ranking. That's a big change in mindset. In safety risk work, teams often ask, "How likely is harm?" In security risk work under TIR57, the question becomes more like, "How feasible is it for someone to exploit this?"
That shift affects the way threat modeling records are written. Required documentation includes:
- System and data flow diagrams
- Trust boundaries
- Exploitability analysis
- Patient-safety impact
- Use-environment assumptions [12]
Those records help show not just that a threat was spotted, but how the device, users, data paths, and setting were evaluated.
Traceability to Controls, Testing, and Residual Risk
TIR57 helps manufacturers connect identified threats to specific security controls and verification evidence. In other words, it ties the risk story to the proof that something was done about it.
It also expands the medical device risk view beyond safety alone by adding another pillar: data and system security. [14] When a security risk can affect patient safety, that risk should be linked to the ISO 14971 safety risk file. That connection is a big deal, because it keeps cybersecurity from sitting off in a separate silo.
TIR57 can also serve as the premarket baseline for TIR97 postmarket management. The structure built before launch carries forward into TIR97's postmarket vulnerability management, which helps teams keep the same line of traceability from early risk analysis through later vulnerability handling.
5. ANSI/AAMI SW96: Medical Device Security - Security Risk Management for Device Software
ANSI/AAMI SW96:2023 lays out a software-focused security risk management process that runs through the full device life cycle. Its main role is simple: it turns threat modeling from a one-off exercise into a documented security risk process.
FDA Recognition or Direct Applicability
FDA recognizes SW96 as a consensus standard that supports QMSR compliance and the cybersecurity duties for cyber devices. That process breaks into four required steps.
Threat Modeling Documentation Requirements
SW96 requires threat modeling to identify threats, security controls, and security design requirements during security risk analysis.[15]
| SW96 Process Element | What It Requires |
|---|---|
| Security Risk Analysis | Threats, vulnerabilities, design requirements |
| Security Risk Evaluation | Assessment strategy, testing results |
| Security Risk Control | Control design, implementation, verification |
| Residual Risk Assessment | Rationale for acceptability |
These outputs feed into the Security Risk Management Report, which serves as the main summary document for the security risk management review.[15] And this paperwork does not end at submission. SW96 keeps these records active after release, so the threat model stays tied to what happens in the field.
Premarket vs. Postmarket Lifecycle Relevance
SW96 covers design, production, and post-production. In plain English, it connects premarket analysis to postmarket vulnerability updates. In the postmarket phase, the threat model must be re-evaluated when the device design changes, when new vulnerabilities are disclosed in third-party components listed in the SBOM, or when routine vulnerability intelligence surfaces new threats.[13]
Traceability to Controls, Testing, and Residual Risk
Under SW96, each threat must map to a control, verification evidence, and a residual-risk decision.[15] That traceability matters because it shows you didn't just spot a risk - you addressed it, tested it, and made a call on what remains.
SW96 also keeps this traceability aligned with safety risk management. It aligns security risk management with ISO 14971:2019 so security controls do not introduce new safety risks.[2][15]
6. ISO 14971: Medical Devices - Application of Risk Management to Medical Devices
Security risk work becomes FDA-relevant when it feeds the safety risk file. ISO 14971 is the main safety-risk standard for cybersecurity threats that could lead to patient harm. Use it to record the safety outcome, not the attack details.
FDA Recognition or Direct Applicability
FDA recognizes ISO 14971 as the core standard for safety risk work. Under QMSR, ISO 14971 records belong in controlled design and risk files, not in separate appendices.
Threat Modeling Documentation Requirements
Keep security risk analysis separate from safety risk analysis, then link them when a cyber threat could affect the patient. The threat model shows the attack path. ISO 14971 records the patient-harm outcome and whether the residual risk is acceptable.
Premarket vs. Postmarket Lifecycle Relevance
Revisit ISO 14971 when postmarket vulnerability intelligence changes the patient-harm profile.
Traceability to Controls, Testing, and Residual Risk
This handoff is the key ISO 14971 record trail. A simple way to think about it: the security team traces how something could go wrong, and the safety file shows what that could mean for the patient.
| Cybersecurity Activity | ISO 14971 / ISO 13485 Link | Documentation Output |
|---|---|---|
| Threat modeling | Design Inputs (ISO 13485 7.3.3) | Security requirements derived from analysis |
| Security risk assessment | Risk Management (ISO 13485 7.1 / ISO 14971) | Security Risk Management Report |
| Residual risk evaluation | Risk Acceptability (ISO 14971) | Residual Risk Record in Safety File |
| Postmarket monitoring | Feedback & Complaints (ISO 13485 8.2.1) | Updated Threat Model and Risk File |
7. AAMI TIR97: Principles for Medical Device Security - Postmarket Risk Management for Device Manufacturers
AAMI TIR97:2019/(R)2023 is the postmarket counterpart to TIR57. It focuses on vulnerability management and residual-risk decisions after a device is released. [16] In plain English, TIR97 takes the premarket threat model and carries it into postmarket monitoring, vulnerability response, and controlled updates.
FDA Recognition or Direct Applicability
FDA recognizes AAMI TIR97 as a postmarket risk-management resource that lines up with its cybersecurity expectations. [9] So the threat model isn't just something you submit once and forget about. It becomes a living postmarket record that should change as the device, its threats, and its use setting change.
Premarket vs. Postmarket Lifecycle Relevance
Postmarket threat models need to reflect the device's actual operating environment, not just the device function on its own. That means looking at the hospital network, connected home setups, Wi-Fi routers, and smartphones that could become attack paths. [1]
The threat model should also be updated as part of the Cybersecurity Management Plan when new vulnerabilities and anomalies show up. [12] A device might look safe on paper at launch, but once it's out in the field, the picture can shift fast.
Threat Modeling Documentation Requirements
TIR97 turns the threat model into something active by linking it to routine monitoring and documented response. Manufacturers are expected to keep a cybersecurity risk report that includes the threat model, risk assessment, SBOM, vulnerability findings, and unresolved anomalies. [12]
A few practical inputs matter here:
- Monitor the CISA KEV catalog to trigger threat-model updates. [12]
- Use SCA, penetration testing, and fuzzing to surface postmarket threats. [1]
This is where postmarket work stops being theory. If a new issue appears in a third-party component or a strange field event starts popping up, the file should show what changed, what was checked, and what the team decided.
Traceability to Controls, Testing, and Residual Risk
Each new vulnerability should be traced from discovery through SBOM impact, control review, and the residual-risk decision. [12][7] That trace matters because it shows whether the issue touched known components, whether current controls still hold up, and whether more action is needed.
When a change stays within scope, manufacturers can use a PCCP to manage approved postmarket updates without a new submission. [1] The postmarket file should also record the patch, user notice, and mitigation. [9]
8. FDA-Recognized NIST and MITRE Threat Modeling Resources
To put those standards into practice, FDA points device makers to MITRE and NIST resources that help turn threat modeling into a repeatable process with clear records. They do not replace FDA expectations. They make the work easier to apply the same way across teams and products. That matters because consistent threat models are easier to use for premarket review and postmarket upkeep.
FDA Recognition or Direct Applicability
The MITRE Playbook for Threat Modeling Medical Devices is the most practical place to start. It was developed with input from FDA and MDIC. A smart approach is simple: use the Playbook to standardize your method, then use NIST publications to back up the analysis and guide control selection.
"The 'Playbook for Threat Modeling Medical Devices' provides a foundation that can inform an organization's threat modeling practices... to develop or evolve an approach to creating threat models in a systematic and consistent way." [17]
FDA also points manufacturers to three NIST publications that support different parts of the model:
| Resource | Best Applied To | Lifecycle Phase |
|---|---|---|
| NIST SP 800-30 | Risk assessment | Early risk analysis |
| NIST SP 800-53 | Controls | Control selection and upkeep |
| NIST SP 800-82 | ICS/robotic devices | Architecture for real-time systems |
Premarket vs. Postmarket Lifecycle Relevance
Each resource fits a different stage of the device lifecycle.
- NIST SP 800-30 works best during early risk analysis.
- NIST SP 800-53 helps with control selection over time, especially for devices tied to cloud backends or enterprise-connected settings.
- NIST SP 800-82 fits actuator-heavy systems, including surgical robotics platforms where command injection is a realistic risk. [9]
Traceability to Controls, Testing, and Residual Risk
These resources help support key decisions around likelihood, impact, control selection, and test depth. Use SP 800-30 to rate risk, SP 800-53 to choose controls, and the MITRE Playbook to shape the threat model itself. Then map each threat to a requirement, a control, and test evidence. [1][9]
This is where a lot of teams stumble. If that chain is weak or missing, FDA often comes back with additional information requests. The point is not to pile on more paperwork. The point is to make the threat model traceable, testable, and maintainable inside the QMS.
Once the model is in place, the next move is to map threats to requirements and evidence.
Threat Modeling Methods That Support FDA-Ready Documentation
Use these methods to turn the standards above into evidence an FDA reviewer can follow.
System and Data Flow Diagrams with Trust Boundaries
System architecture diagrams and data flow diagrams (DFDs) show the device, hospital or clinic networks, home settings, phones and other connected devices, and software update servers. They also show the interfaces and dependencies between them.
Trust boundaries mark the points where data moves between areas with different levels of access or different owners. That matters because those crossings are often where risk shows up first. These diagrams also give you the traceability FDA looks for, from system scope all the way to risk control.
FDA expects threat modeling to go beyond the device by itself and reflect the larger system around it.[1][2] That bigger-picture view is what FDA wants to see in premarket submissions.
STRIDE and Scenario-Based Threat Enumeration
Once the architecture is mapped, STRIDE gives teams a clear way to list threats. Attack trees then help show how one threat scenario might play out across an attack path.
Used together, they answer a simple but important question: how could this threat get to the device? That makes the analysis easier to tie back to design choices, controls, and test work.
SBOM-Informed Component Analysis
Under Section 524B of the FD&C Act, manufacturers of cyber devices must provide a Software Bill of Materials (SBOM). Use the SBOM to identify third-party and open-source dependencies for software composition analysis (SCA).[3]
SCA helps identify inherited vulnerabilities in those components so teams can account for supply chain risk as part of the security analysis.[1]
Using MITRE and NIST Resources in Practice
Use MITRE and NIST resources to standardize threat names, mitigation mapping, and residual-risk records. When manufacturers use these resources the same way across the process, the threat model becomes easier to trace through requirements, controls, and verification evidence.
Known vulnerabilities and weak controls are foreseeable failure modes, so document and address them directly.
Next, map each threat to requirements, controls, and verification evidence.
Turning Threat Models Into Submission-Ready Evidence
Map Threats to Requirements, Controls, and Test Evidence
Once the threat model is set, the next step is to turn it into FDA evidence you can trace from start to finish. A threat model is ready for FDA review when each threat maps to a requirement, a control, a test, and a residual-risk rationale.
Use exploitability-based scoring instead of historical likelihood estimates. Document the attack vector, attack complexity, and privileges required.
Here’s what each mapping element should show:
| Mapping Element | What to Document |
|---|---|
| Threat Scenarios | Identify which threats exploit specific vulnerabilities, using STRIDE or another structured method |
| Security Requirements | Define the design requirements intended to mitigate identified threats |
| Controls | Map controls to the eight Appendix 1 categories, such as Authentication, Authorization, and Cryptography |
| Verification Testing | Link each control to test reports such as penetration testing, fuzzing, and vulnerability scans |
| Residual Risk | Document an exploitability-based rationale for why the remaining risk is acceptable |
If a vulnerability isn’t resolved, it needs a documented risk acceptance or a by-design rationale. No loose ends.
Structure Evidence for FDA Premarket Submissions
That traceability falls apart if the submission package uses a different logic than the threat model. The filing structure has to mirror the evidence trail.
FDA’s eSTAR system requires cybersecurity deliverables to go into the required attachment slots, including the threat model, SBOM, and postmarket cybersecurity management plan [5]. If that documentation is missing, the result can be a Technical Screening hold or a Refuse to Accept (RTA) decision. FDA has used that authority since October 2023 [6].
The submission package should cover:
- hostile environment assumptions
- a global system view that shows how the device fits into the broader network and data ecosystem
- update security, backed by test evidence for digital signature verification and rollback procedures
- a machine-readable SBOM in CycloneDX or SPDX JSON format, plus a vulnerability assessment for each listed component
- decommissioning-phase risks documented directly in the threat model [5][7]
Keep the threat model, SBOM, and risk record under configuration control so changes stay traceable through design inputs and corrective action.
With the evidence organized, the next step is keeping that material controlled across the product lifecycle.
Conclusion
Taken together, these standards make threat modeling a required engineering control across the full product lifecycle. It is now a regulated, lifecycle-wide requirement, not a one-off security task.
Key Takeaway for Manufacturers
The main value of threat modeling is not compliance paperwork. It is disciplined design control.
The gap, in practice, comes from treating threat modeling like a document to file away instead of a control that shapes the product. Cybersecurity still shows up often as a deficiency in FDA submissions, and the way to close that gap is to build threat modeling into design inputs, security controls, verification testing, and postmarket monitoring.
What a Strong FDA-Aligned Program Looks Like
A strong program connects threats to design inputs, verified controls, and postmarket updates through the QMS. FDA guidance, QMSR, ISO 14971, SW96, TIR57, and TIR97 should work as one traceable system.
Premarket analysis should feed design controls. Postmarket monitoring should feed updated risk records. That is the FDA-ready posture: traceable before submission, current after release, and controlled inside the QMS.
FAQs
What counts as a complete FDA-ready threat model?
A complete, FDA-ready threat model is a diagram-driven analysis. It’s not just a bulleted list.
It needs to cover the entire medical device system, including hospital networks, cloud infrastructure, and update mechanisms.
The model should document:
- Architecture and data flows
- Trust boundaries
- The chosen method
- How the model connects to design decisions, supply chain risks, security controls, residual risks, and environmental assumptions
In plain terms, the threat model should show how the system works, where the risk sits, and why certain security choices were made. That link matters. If a design decision affects risk, the model should make that easy to see.
How often should a medical device threat model be updated?
The FDA expects threat modeling to be an iterative, continuous process across the medical device lifecycle, not a one-time task.
That means manufacturers should revisit and update the threat model as cybersecurity risk analysis changes, especially when the device is developed further, deployed in new settings, or affected by changes in the connected systems around it.
How does threat modeling connect to the SBOM and QMS?
Threat modeling, the SBOM, and the QMS all play a linked role in the FDA’s secure-by-design framework.
Threat modeling lays out the system’s security architecture and points to where risk may show up. The SBOM then lists the software components, which helps teams spot where those risks exist in the code and dependencies.
Under the QMSR, both need to sit inside controlled quality processes. Threat models should tie back to design controls so teams can trace security decisions to product development. The SBOM supports vulnerability assessments by mapping software dependencies to known threats.
