FDA Guidance: Securing Third-Party Libraries in Devices
Post Summary
The FDA has tightened its focus on cybersecurity risks in medical devices, particularly those arising from third-party software libraries. These components - whether open-source, commercial, or off-the-shelf - often make up the majority of a device's codebase and are critical for functions like encryption, connectivity, and data processing. However, they also introduce significant vulnerabilities that can compromise patient safety.
Key takeaways from the FDA's updated guidance include:
- Mandatory Software Bill of Materials (SBOM): Every device must include a detailed, machine-readable SBOM, covering all third-party components.
- Continuous Vulnerability Monitoring: Manufacturers must actively track and address security flaws in third-party code throughout the device's lifecycle.
- Timely Patching: Prompt updates are required to resolve vulnerabilities, reducing risks to patient safety.
- Integration into Quality Systems: Starting February 2, 2026, cybersecurity requirements for third-party libraries must align with ISO 13485 standards, including design and purchasing controls.
With cybersecurity incidents in medical devices rising sharply (e.g., a 437% increase in remote code execution exploits in 2023), these measures are not optional. Tools like automated SBOM management platforms can help manufacturers meet these requirements efficiently, ensuring compliance and safeguarding healthcare systems.
FDA Cybersecurity Expectations for Third-Party Libraries

What Are Third-Party Libraries in Medical Devices?
Third-party libraries are external software components integrated into a medical device's software. These libraries typically fall into three categories: commercial software (licensed products), open-source components (publicly available code), and off-the-shelf (OTS) software (ready-made packages used without modification). The FDA also uses the term Software of Unknown Provenance (SOUP) to describe third-party components that lack a well-documented development history or security assessment.
These libraries often handle essential functions like encryption, network communication, image processing, and protocol operations, playing a critical role in the device's functionality. Under Section 524B(c) of the FD&C Act, a device is classified as a cyber device if it includes software, connects to the internet (via Wi-Fi, Bluetooth, or USB), and has characteristics that make it susceptible to medical device security risks [2]. This definition encompasses most modern connected medical devices.
Key FDA Guidance Documents and Regulations
The FDA has issued several regulations and guidance documents to ensure manufacturers properly manage third-party libraries:
| Regulation / Guidance | Scope | Requirements for Third-Party Libraries |
|---|---|---|
| FD&C Act Section 524B | Statutory law | Requires an SBOM for all commercial, open-source, and OTS components; mandates ongoing vulnerability monitoring. |
| QMSR (ISO 13485:2016) | Quality system | Includes Clause 7.4, which introduces purchasing controls for cybersecurity-critical suppliers, effective February 2, 2026. |
| Premarket Cybersecurity Guidance | Submission content | Requires documentation under a Secure Product Development Framework (SPDF) and threat modeling for third-party interfaces. |
| Postmarket Cybersecurity Guidance | Lifecycle management | Establishes coordinated vulnerability disclosure (CVD) processes for third-party components. |
| 21 CFR 820.30 (Design Controls) | Device design | Requires design inputs and outputs to address all components, including third-party software. |
The 2023 premarket cybersecurity guidance, updated in February/March 2026, emphasizes integrating third-party library security within a broader Secure Product Development Framework (SPDF). Starting October 1, 2023, the FDA began holding eSTAR submissions if cybersecurity documentation, including third-party component details, is incomplete or inaccurate. These updates, combined with the QMSR requirements effective February 2, 2026, highlight the importance of addressing third-party library security from the outset.
FDA Security Goals as They Apply to Third-Party Libraries
The core aim of these regulations is to ensure device safety by securing third-party libraries. To meet the FDA's standards for "reasonable assurance of safety and effectiveness" [2], manufacturers must focus on three critical areas outlined in Section 524B:
- Comprehensive SBOM: Maintain a complete Software Bill of Materials (SBOM) that includes all commercial, open-source, and OTS components.
- Continuous Vulnerability Monitoring: Implement a plan to monitor vulnerabilities throughout the device's entire lifecycle.
- Timely Patching: Deploy patches promptly to address vulnerabilities in third-party components, ensuring devices remain secure by design [2].
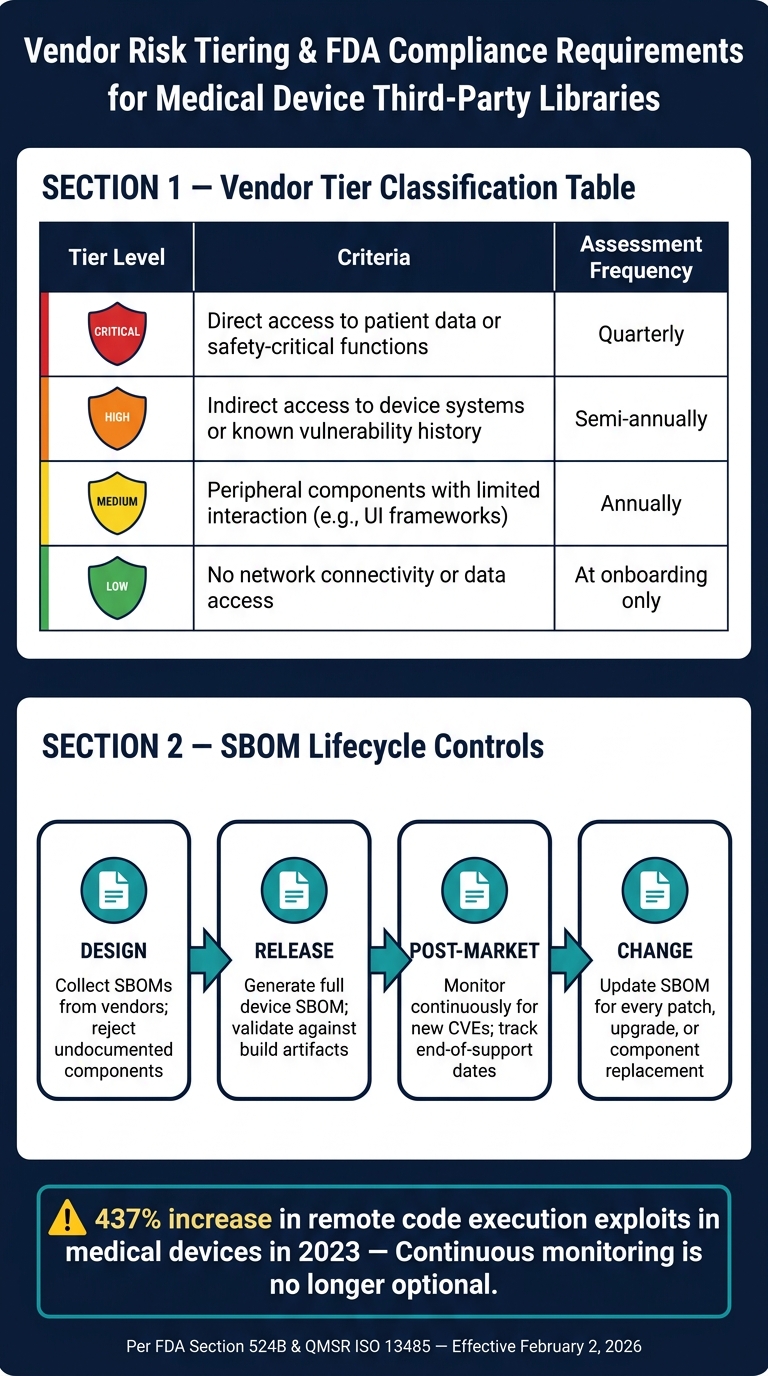
The urgency of these measures is underscored by a 437% increase in remote code execution and privilege escalation exploits in medical devices in 2023 [1]. Such statistics highlight the direct connection between third-party library security and patient safety.
sbb-itb-535baee
Practical Guide to Cybersecurity and SBOM Management for FDA Approval
Aligning Third-Party Library Security With FDA Compliance
FDA Cybersecurity Requirements for Third-Party Libraries in Medical Devices
Mapping Security Practices to FDA Requirements
Meeting FDA expectations requires actionable steps, starting with your Software Bill of Materials (SBOM). As previously mentioned, the SBOM must document every third-party component and be submitted in a machine-readable format. The FDA recognizes SPDX and CycloneDX as acceptable standards. Keep your SBOM up to date, reflecting all changes to ensure accuracy. This serves as the backbone for security testing and vendor assessments.
Security testing should cover the entire system, not just your proprietary code. This includes vulnerability scanning, penetration testing, and fuzz testing for third-party libraries and SOUP components. Any vulnerabilities identified must be processed through your Corrective and Preventive Action (CAPA) system. This ensures alignment with the FDA's updated QMSR regulations under ISO 13485, which take effect on February 2, 2026.
Vendor oversight should follow a tiered classification system:
| Vendor Tier | Criteria | Assessment Frequency |
|---|---|---|
| Critical | Direct access to patient data or safety-critical functions | Quarterly |
| High | Indirect access to device systems or known vulnerability history | Semi-annually |
| Medium | Peripheral components with limited interaction (e.g., UI frameworks) | Annually |
| Low | No network connectivity or data access | At onboarding |
Contracts with vendors should include cybersecurity clauses, such as mandatory notifications within 24 hours for critical vulnerabilities and the requirement to provide SBOMs in SPDX or CycloneDX formats. As global MedTech expert Ran Chen aptly states:
"Every connected medical device is only as secure as its weakest supplier." [1]
To meet FDA premarket standards, these testing and oversight practices must be paired with strong documentation.
Documentation and Premarket Submission Requirements
Premarket submissions must demonstrate that third-party library security is integrated throughout the development process.
Key documentation includes a machine-readable SBOM, a threat model that addresses risks from third-party components and communication interfaces, and comprehensive security testing results for the entire device. Your ISO 14971 risk analysis should treat vulnerabilities in vendor components as potential hazards. Additionally, traceability between third-party security features and your design outputs is required under 21 CFR 820.30 design controls.
For legacy devices, the FDA expects manufacturers to inventory all third-party components, even if vendor support has ended. When patches are unavailable, compensating controls - like network segmentation - must be documented. You must also account for Nth-party risks, such as issues stemming from your vendors' sub-suppliers (e.g., a cloud provider's infrastructure partner), in your submission. Addressing these risks signals a proactive stance toward FDA compliance.
"Under QMSR, cybersecurity requirements must be included in purchasing specifications for cybersecurity-critical components and services." - Ran Chen, Global MedTech Expert [1]
If your device uses third-party AI/ML libraries, documentation must also cover risks like model poisoning or adversarial manipulation of training data. These are areas of growing interest for the FDA, especially as AI-enabled devices become more prevalent.
Building a Secure Product Development Framework (SPDF)
A Secure Product Development Framework weaves security measures into every stage of your device's lifecycle, starting from its design phase and extending through post-market monitoring. The FDA's 2026 guidance underscores that third-party library security must be integrated into your existing Quality Management System (QMS). This is not something to treat as a separate IT issue.
Governance and Risk Assessment for Third-Party Libraries
Strong governance, based on documented security practices, is critical for managing risks tied to third-party libraries. This begins by addressing third-party libraries as potential hazard sources within your ISO 14971 risk analysis. Under the updated QMSR, effective February 2, 2026, oversight of these libraries must be embedded into ISO 13485 Clause 7.4 (Purchasing Controls) and Clause 7.3 (Design Controls). Relying on informal processes outside the QMS is no longer sufficient.
One way to strengthen governance is by implementing a pre-integration rejection policy. For example, disqualify any third-party library that lacks proper security documentation or a machine-readable Software Bill of Materials (SBOM). For vendors you do accept, request clear evidence across these critical areas:
| Assessment Dimension | Evidence to Request |
|---|---|
| Security Certifications | ISO 27001, SOC 2 Type II, or IEC 62443 audit reports |
| Vulnerability Management | Disclosure policy, mean time to patch, and CVE history |
| Secure Development | SDLC documentation and penetration test reports |
| Supply Chain Transparency | The vendor's SBOM practices and sub-tier oversight |
Equally important are contractual obligations. Without enforceable agreements, governance has limited impact. Contracts should mandate that vendors notify you of vulnerabilities within 24 hours and high-risk issues within 72 hours [1]. They should also require vendors to deliver SBOMs in standardized machine-readable formats like SPDX or CycloneDX.
Lifecycle Controls for Third-Party Libraries
Even a library deemed low-risk at the time of onboarding can become vulnerable later due to new CVEs. This makes it essential to maintain lifecycle controls throughout the entire lifespan of the device - from design to retirement.
Here’s a simplified checklist for managing SBOMs across various lifecycle stages:
| SBOM Lifecycle Stage | Action Required |
|---|---|
| Design | Collect SBOMs from vendors; reject components without documentation |
| Release | Generate a complete device SBOM; validate it against build artifacts |
| Post-market | Monitor continuously for new CVEs; track end-of-support dates |
| Change | Update the SBOM for every patch, version upgrade, or component replacement |
For legacy devices, it’s crucial to inventory outdated libraries and document compensating controls, such as network segmentation. Regulators often flag end-of-life components as a significant gap during premarket reviews.
These lifecycle controls provide a foundation for leveraging tools that automate risk monitoring.
Using Platforms to Manage Third-Party Library Risk
Managing third-party library risks manually - using spreadsheets and annual reviews - is no longer practical. In 2023, remote code execution and privilege escalation exploits in medical devices surged by 437% [1]. This sharp increase highlights the inadequacy of periodic assessments and the growing need for continuous monitoring.
Platforms specifically designed for this purpose can make a real difference. For instance, Censinet RiskOps™ is tailored for healthcare organizations, offering streamlined third-party risk assessments, continuous vendor monitoring, and collaborative risk management. Its Censinet AITM™ feature speeds up the assessment process by allowing vendors to complete security questionnaires almost instantly and automatically summarizing their evidence. This reduces the time spent on manual reviews while keeping human oversight intact. For medical device manufacturers juggling multiple third-party library vendors across various product lines, this type of automation aligns with the continuous monitoring requirements outlined in FDA Section 524B and the 2026 QMSR.
Managing SBOMs and Dependency Tracking
Staying on top of Software Bill of Materials (SBOM) management and tracking dependencies is essential for aligning third-party library practices with FDA cybersecurity guidance.
The Role of SBOMs in FDA Compliance
SBOM management plays a central role in meeting FDA requirements under the secure product development framework. Since March 2023, Section 524B of the FD&C Act mandates that any "cyber device" seeking premarket clearance must include an SBOM in its submission. The FDA broadly defines a cyber device as any device running software and connecting via interfaces like Wi-Fi, Bluetooth, or USB. Starting October 1, 2023, the FDA can reject premarket submissions lacking sufficient cybersecurity details, including SBOMs [3].
"An incomplete inventory is now grounds for refusal, and there's no ambiguity in the current framework about that." - Christian Espinosa, Founder & CEO, Blue Goat Cyber [3]
Your SBOM must detail all software components - proprietary code, open-source libraries, third-party packages, and commercial off-the-shelf (COTS) software - along with their dependency trees [3][4]. It must also be machine-readable (e.g., CycloneDX, SPDX) for FDA review. CycloneDX is often preferred for its built-in Vulnerability Exploitability eXchange (VEX) support and Common Vulnerabilities and Exposures (CVE) integration [3].
Each entry in the SBOM should include the NTIA baseline fields: supplier name, component name, version, unique identifier (like PURL or CPE), dependency relationships, author, and timestamp. Additionally, the FDA expects lifecycle metadata, which should indicate whether a component is actively maintained, in legacy status, or unsupported, along with its end-of-support (EOS) date [3][4].
Next, let’s look at how to keep your SBOMs updated throughout the lifecycle of your device.
Keeping SBOMs Current
Automating SBOM generation within your CI/CD pipeline ensures your inventory stays updated with every patch, dependency update, or component removal. Tools like Syft or Yocto’s create-spdx plugin can automatically generate SBOMs during each build. To maintain accuracy, validate the SBOM against build artifacts before releasing updates. For legacy devices without source code access, binary analysis tools can identify third-party components and flag potential risks [3].
Using SBOMs to Monitor for Vulnerabilities
An updated SBOM is the foundation for effective vulnerability monitoring, which is essential for FDA compliance. Use it to continuously cross-check against resources like the National Vulnerability Database (NVD), CISA’s Known Exploited Vulnerabilities (KEV) Catalog, and vendor security advisories. Document and evaluate every CVE you identify.
Not all CVEs in third-party libraries will impact your specific device configuration. VEX statements let you formally communicate to the FDA and customers when a CVE is irrelevant to your device, helping focus remediation efforts where they’re needed most [4]. For exploitable vulnerabilities, include them in your ISO 14971 risk analysis and address them through your CAPA system, treating them as potential hazard sources.
For components approaching their EOS date, plan migrations or implement compensating controls, like network segmentation, and document the associated risks and mitigation strategies. Regulators often flag unsupported components during reviews, and with remote code execution exploits in medical devices rising 437% in 2023 [1], delaying remediation is no longer an option.
Healthcare organizations can simplify these processes by combining SBOM management and vulnerability tracking within platforms like Censinet RiskOps™. This approach supports continuous FDA compliance while strengthening the cybersecurity of medical devices. Together, these practices provide a solid framework for regulatory adherence and long-term device security.
Conclusion and Key Takeaways
Ensuring the security of third-party libraries in medical devices isn't just a best practice - it's a legal requirement. With FDA Section 524B and the QMSR taking effect on February 2, 2026, manufacturers must apply the same stringent standards to open-source packages, COTS components, and vendor-supplied libraries as they do to proprietary code. The urgency is underscored by alarming statistics: remote code execution exploits in medical devices jumped by 437% in 2023, and healthcare data breaches cost an average of $9.8 million in 2024 [1].
To stay compliant and protect patient safety, manufacturers should focus on these key practices:
- Maintain an up-to-date, machine-readable SBOM (in CycloneDX or SPDX format) with every build.
- Categorize vendors by risk level - critical suppliers, especially those handling patient data, should undergo quarterly assessments, while lower-risk components can be reviewed annually.
- Incorporate cybersecurity into your QMS, ensuring vulnerabilities tied to vendors are addressed through the CAPA process under ISO 13485 Clause 7.4.
- For legacy devices that can't be patched, deploy compensating controls like network segmentation and thoroughly document associated risks.
Managing these responsibilities across numerous suppliers and components is no small task. Tools like Censinet RiskOps™ can streamline processes such as vendor tiering, SBOM validation, and ongoing vulnerability monitoring. These platforms help maintain compliance without losing the human oversight essential for safeguarding patient safety.
The FDA's expectations are clear, and the risks are significant. Security must be embedded into the supply chain from the start, rather than being patched together after an incident. By following the regulatory guidance and risk management strategies outlined here, manufacturers can consistently meet both legal requirements and patient safety expectations.
FAQs
Do I need an SBOM for every device update?
Yes, the FDA mandates that Software Bills of Materials (SBOMs) be updated with every software release. These updates must precisely represent the device's components. To achieve this, it's crucial to regenerate the SBOM during each build process to match the final artifact. Maintaining these updated versions is key for managing risks effectively and creating a reliable audit trail throughout the device's lifecycle.
How can I prove a CVE doesn’t impact my device?
Pairing your Software Bill of Materials (SBOM) with a Vulnerability Exploitability eXchange (VEX) document helps you determine if a flagged CVE in your device is actually exploitable. To take this a step further, use reachability analysis tools to check if the vulnerable code is active within your system. This approach ensures you're addressing real risks instead of chasing false alarms. Tools like Censinet RiskOps make it easier to manage these evaluations throughout the entire product lifecycle.
What should I do when a legacy device can’t be patched?
If a legacy device can't be patched, the first step is to reach out to the manufacturer to discuss possible compliance solutions. Perform a thorough vulnerability assessment to pinpoint risks and evaluate how they might affect the device's safety. Once risks are identified, put compensating controls and mitigation strategies in place to address security concerns. Tools like Censinet RiskOps™ assist healthcare organizations in simplifying these steps, helping to manage risks effectively across medical devices and clinical applications.

