Medical devices need cybersecurity built into their design - not added later. The FDA's secure design framework prioritizes patient safety by embedding security into every stage of a device's lifecycle. This approach contrasts sharply with older, reactive methods that rely on perimeter defenses and post-launch fixes. These legacy approaches often leave devices vulnerable to critical medical device security risks that threaten patient safety. Key differences include:
- FDA Secure Design: Integrates cybersecurity from the start, focusing on safety and device functionality.
- Older Methods: Emphasize external defenses and fixing vulnerabilities after deployment.
Since 2023, the FDA has enforced strict requirements, including mandatory cybersecurity documentation and Software Bill of Materials (SBOM). By 2026, manufacturers must align with the Quality Management System Regulation (QMSR), ensuring security is part of broader quality processes.
Quick Takeaways:
- Secure design reduces long-term costs by addressing vulnerabilities early.
- Traditional methods often fail to safeguard devices used for 10–20 years.
- Manufacturers must now provide detailed, traceable security plans for FDA approval.
This shift ensures devices remain safe for patients while meeting evolving regulatory standards.
Webinar: Master Medical Device Cybersecurity: Avoid FDA Delays
sbb-itb-535baee
FDA Secure Design Principles: A Safety-First Framework
The FDA's secure design framework places cybersecurity at the core of every phase in the lifecycle of medical devices, recognizing that security risks directly impact patient safety.
The Secure Product Development Framework (SPDF)
The Secure Product Development Framework (SPDF) is the FDA's recommended approach for embedding cybersecurity throughout a product’s entire lifecycle - from the initial concept to its eventual decommissioning. This framework is designed to align seamlessly with existing Quality Management Systems (QMS). Starting February 2, 2026, under the QMSR (which aligns with ISO 13485:2016), manufacturers will be required to document their systematic security activities in the Design History File.
"Security should be properly built into the product throughout development instead of being 'bolted on' at the end after testing." - Brett Todd, Director – Systems Engineer, Full Spectrum
Key Secure Design Techniques
The SPDF builds on established engineering practices, beginning with threat modeling. This process maps out how potential attackers might interact with a device and identifies possible clinical harms and enterprise risks. Importantly, this analysis happens early - before substantial code is written - ensuring that security requirements guide the product's architecture from the outset.
Following threat modeling, manufacturers apply techniques such as attack surface reduction, secure boot, encryption for both data in transit and at rest, and secure update mechanisms to prevent unauthorized software modifications. A unique aspect of SPDF’s approach is its environment-aware analysis, which evaluates devices in real-world settings like hospital Wi-Fi networks and interconnected clinical systems.
Once these measures are in place, structured testing ensures the security features function as intended.
Verification and Validation Requirements
Building security into a product isn’t enough - it must also be rigorously tested. Under the SPDF, verification and validation (V&V) go beyond a single penetration test. Instead, manufacturers must use a combination of testing methods, including penetration testing, fuzz testing, and static/dynamic code analysis. These tests collectively confirm both the safety and effectiveness of the security measures.
A critical component of the process is the Software Bill of Materials (SBOM), a machine-readable inventory of all software components within the device. As mandated by Section 524B of the FD&C Act, cyber devices must include an SBOM. This allows manufacturers to quickly identify which products might be affected when new vulnerabilities are discovered - a crucial capability, especially since the average cost of a healthcare data breach reached $9.8 million in 2024.
Premarket submissions must also include outputs from threat modeling and a vulnerability management plan. Many manufacturers use automated vendor solutions to streamline these assessments. These must be traceable to specific design decisions documented in the Design History File. The FDA places a strong emphasis on this traceability during inspections, particularly under the Compliance Program Manual (#7382.850), which took effect on February 2, 2026.
| V&V Activity | Purpose | FDA Expectation |
|---|---|---|
| Penetration Testing | Simulate real-world attacks on the device | Demonstrate exploitability in the intended environment |
| Fuzz Testing | Identify unexpected input-handling failures | Catch defects early as part of an automated pipeline |
| Static/Dynamic Code Analysis | Identify vulnerabilities in source and runtime code | Integrate into the development workflow |
| SBOM Provision | Inventory all software components | Required for cyber devices under Section 524B |
| Vulnerability Management Plan | Address post-release vulnerabilities | Include in premarket submissions; maintain postmarket |
Traditional Cybersecurity Approaches: Where They Work and Where They Fall Short
Traditional cybersecurity methods are well-suited for IT environments like corporate networks and data centers. But when applied to medical devices, their limitations become glaring.
Perimeter-Focused Security
The traditional IT security model relies on firewalls, VPNs, and intrusion detection systems to create a secure boundary around the network. The idea is simple: keep threats out, and the systems inside will stay safe. However, medical devices often don’t fit neatly within this perimeter. Many rely on wireless communication protocols, which can be exploited - even if the device isn’t designed to connect to the internet [6]. While network segmentation can reduce the impact of breaches, it doesn’t address vulnerabilities baked into the device’s design.
Reactive Hardening and Patch Management
Traditional IT security tends to respond to threats as they arise: a vulnerability is identified, a patch is developed, and updates are deployed. While this works for laptops and servers, it’s far less effective for medical devices. Fixing security flaws in devices after deployment can cost 4 to 5 times more than addressing them during the design phase [7]. For older devices, patching may not even be feasible due to technical limitations, forcing manufacturers to rely on alternative safeguards. Compounding this challenge, Section 524B of the FD&C Act now requires manufacturers to maintain a documented plan for monitoring and addressing vulnerabilities after devices are on the market [3]. This marks a major departure from the traditional "patch as needed" mindset, highlighting the need for a more proactive, safety-focused approach in medical device cybersecurity.
IT-Centric Risk Orientation
Traditional IT security frameworks revolve around the CIA triad: Confidentiality, Integrity, and Availability. The primary goal is to protect data from unauthorized access. Medical device cybersecurity, however, prioritizes patient safety and essential performance - ensuring devices operate correctly and remain available to prevent harm. This difference highlights the limits of conventional IT security when applied to healthcare. As George Strom, Director at Intertek Connected World, explains:
"A connected device that performs clinically but fails under cybersecurity stress is not meeting intended use expectations in today's regulatory environment." [2]
This distinction is crucial. A technical flaw in a medical device isn’t just a data problem - it can directly threaten patient safety.
FDA Secure Design vs. Traditional Cybersecurity: A Direct Comparison
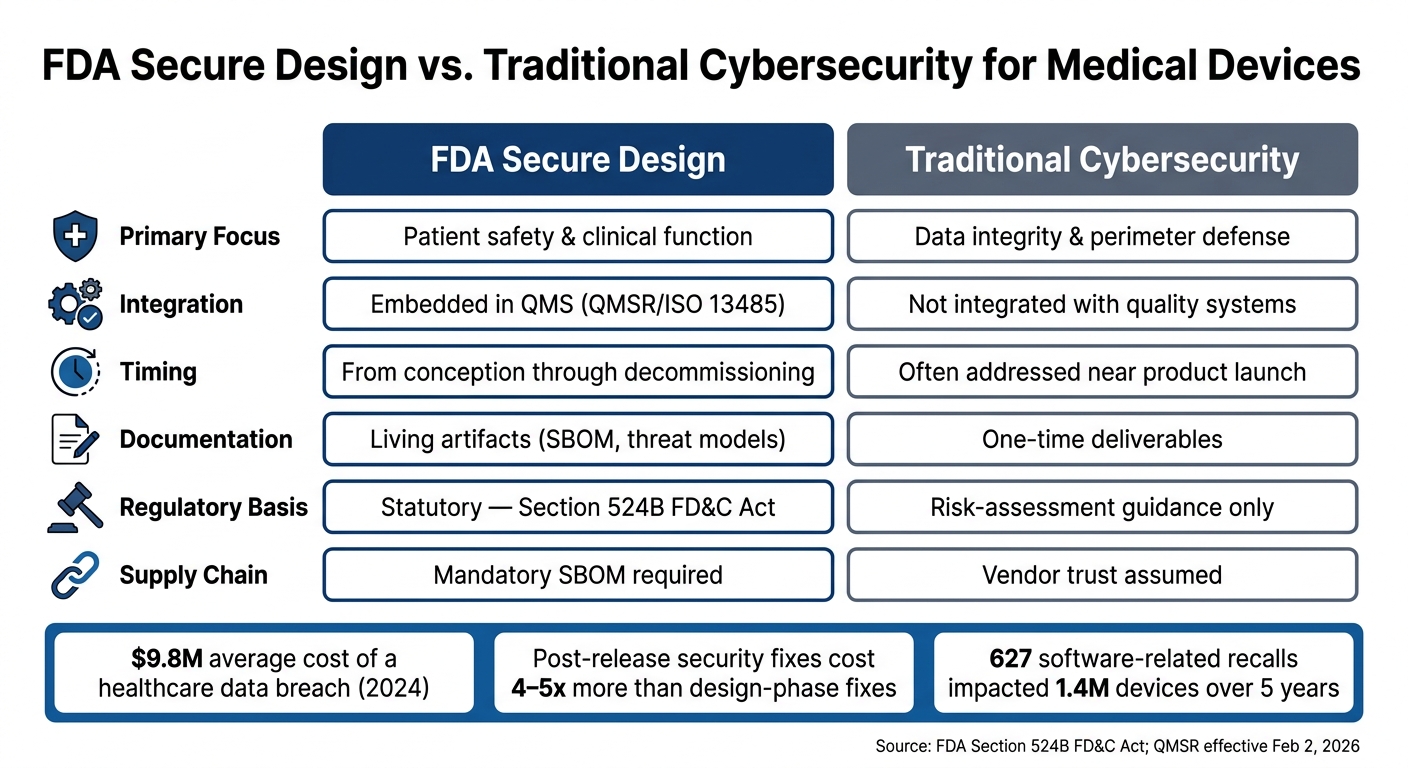
FDA Secure Design vs. Traditional Cybersecurity: Key Differences for Medical Devices
Differences in approach influence FDA review processes and, ultimately, patient safety.
Regulatory Alignment and Documentation Requirements
The FDA's approach to cybersecurity goes beyond traditional one-time reporting. It requires continuous documentation through controlled Quality Management System (QMS) processes. Under Section 524B of the FD&C Act, cybersecurity compliance is now a statutory requirement. Since October 1, 2023, the FDA has been authorized to issue "Refuse to Accept" holds on premarket submissions - such as 510(k), PMA, De Novo, and HDE - that fail to meet specific cybersecurity standards [3].
By February 2, 2026, the Quality Management System Regulation (QMSR) - aligned with ISO 13485:2016 - became the permanent framework for medical device quality systems [1]. A Software Bill of Materials (SBOM) is no longer just an appendix; it's a dynamic document integrated into QMS processes and the Design History File.
"The difference between programs that move through FDA review cleanly and those that encounter friction usually isn't technical capability. It's whether cybersecurity was treated as an architecture decision early on or as a documentation exercise closer to submission." - Matt Rengo, Lead, Punch Through [5]
This emphasis on documentation supports better lifecycle management and regulatory readiness.
Safety-Driven vs. Perimeter-Driven Design Philosophy
The FDA's secure design philosophy diverges sharply from traditional IT cybersecurity approaches. Here's a breakdown of the fundamental differences:
| Feature | FDA Secure Design (SPDF) | Traditional Cybersecurity |
|---|---|---|
| Primary Focus | Patient safety & clinical function | Data integrity & perimeter defense |
| Integration | Embedded in QMS (QMSR/ISO 13485) | Not integrated with quality systems |
| Timing | From conception through decommissioning | Often addressed near product launch |
| Documentation | Living artifacts (SBOM, threat models) | One-time deliverables |
| Regulatory Basis | Statutory (Section 524B FD&C Act) | Risk-assessment guidance |
| Supply Chain | Mandatory SBOM | Vendor trust |
Traditional cybersecurity often asks, "Is the network perimeter secure?" FDA secure design takes a broader view, asking harder questions. Naomi Schwartz of Medcrypt explains:
"The FDA is driving people to think, 'How much harm could I do if I caused 10% of people with a given condition to have a serious medical emergency that overwhelms the hospital system?' That's a public health problem." [1]
This shift - from focusing on network security to addressing public health risks - is at the heart of the FDA's approach. It ensures patient safety is embedded into every design decision.
Lifecycle Management and Postmarket Surveillance
Traditional IT security typically applies patches as needed, but the FDA's model requires a more comprehensive approach. Security obligations now span the entire product lifecycle, starting at the proof-of-concept stage and extending through decommissioning.
Under Section 524B, manufacturers must maintain a documented plan for monitoring and addressing postmarket vulnerabilities. This includes two tiers of patching: a "reasonably justified regular cycle" for known vulnerabilities and immediate out-of-cycle patches for critical risks that could harm patients [4]. For legacy devices, Section 524B doesn't apply retroactively to devices cleared before March 29, 2023. However, any new premarket submission for a modification triggers compliance with the latest cybersecurity requirements [3].
The stakes are high. Fixing a software security flaw after a product is released can cost 4 to 5 times more than addressing it during the design phase [7]. Proactive lifecycle planning not only ensures compliance but also reduces costs and enhances device safety - making it a smart move for manufacturers.
Putting FDA Secure Design Into Practice in Healthcare Settings
Embedding Secure Design into Product Development
Cybersecurity in manufacturing needs to be treated as an integral part of the design process, not just a box to check for submission. This means weaving the Secure Product Development Framework (SPDF) directly into your ISO 13485 quality system, rather than running it as a separate initiative.
To make this work, tie every security-related artifact to a specific Quality Management System (QMS) clause. For example:
- Threat models align with Design Inputs (Subclause 7.3.3)
- Security testing corresponds to Design Verification (7.3.7)
- Vulnerability handling links to Corrective Action (8.5.2)
By doing this, documentation becomes a natural part of the development process, avoiding the stress of scrambling to compile it just before submission. This approach also strengthens risk management as the project progresses.
"The key is to make documentation a natural byproduct of your development work, rather than a separate, painful task you save for the end." - Henry Anfang, Firmware Developer, Punch Through [8]
During the design phase, applying the S.T.R.I.D.E. methodology - which addresses Spoofing, Tampering, Repudiation, Information Disclosure, Denial of Service, and Elevation of Privilege - ensures proactive security measures are baked into the product. Retrofitting security into an already-developed architecture is far more challenging. For teams implementing their first SPDF, the process typically takes 10 to 14 weeks of concentrated effort [10].
Manufacturers that follow a structured and repeatable SPDF process have seen consistent success. A case in point is Blue Goat Cyber, which reports a 100% acceptance rate across 250+ FDA submissions [10]. This demonstrates the value of disciplined and well-documented processes.
But internal practices alone aren't enough. Managing external components is equally crucial to ensure comprehensive device security.
Managing Third-Party and Supply Chain Risks
Third-party components often introduce hidden vulnerabilities into medical devices. For instance, over five years, 627 software-related recalls impacted 1.4 million units due to issues like unpatched open-source libraries [7].
A key tool for managing these risks is a living SBOM (Software Bill of Materials). Instead of generating it once for submission, treat it as a dynamic artifact within your QMS, updated with every build. Tools like Syft can automate this process by integrating directly into CI/CD pipelines. Build gates can be set to automatically fail if a new library introduces a critical vulnerability [8].
For vulnerabilities flagged but deemed non-impactful to the device, Vulnerability Exploitability eXchange (VEX) documents can be used to formally explain why a component is "not_affected." This reduces unnecessary noise and ensures focus remains on critical issues [8].
If a third-party supplier cannot provide an SBOM, document the gap with a written justification and conduct a dedicated risk analysis for those components. Additionally, postmarket monitoring should include tracking the CISA Known Exploited Vulnerabilities (KEV) catalog to catch emerging threats before they pose risks to patient safety [9].
By combining secure internal development practices with rigorous oversight of third-party components, manufacturers can meet FDA's expectations for safeguarding patient safety throughout a device's lifecycle.
Using Cyber Risk Management Platforms to Support Compliance
Managing both internal and supply chain risks adds layers of operational complexity, especially for healthcare delivery organizations (HDOs) and manufacturers juggling FDA cybersecurity requirements across multiple devices and vendors.
Platforms like Censinet RiskOps™ are designed to simplify these challenges. This tool streamlines third-party and enterprise risk assessments, facilitates cybersecurity benchmarking, and supports collaborative risk management across medical devices, clinical applications, and supply chains.
One standout feature is Censinet AI™, which speeds up the assessment process by enabling vendors to complete security questionnaires in seconds. It automatically summarizes evidence, generates risk reports, and reduces the time spent on manual review - all while keeping human oversight in critical decisions. For HDOs managing numerous vendors, this balance of efficiency and control is invaluable in maintaining the continuous compliance FDA requires for postmarket surveillance.
Conclusion: Moving Medical Device Cybersecurity Forward
Key Takeaways for Medical Device Manufacturers
Traditional approaches to cybersecurity often treat it as something to be added later, but FDA's secure design philosophy demands that security be built in from the very beginning. Why does this matter? Fixing security flaws after a product is released is 4–5 times more expensive. Over the past five years, there have been 627 recalls, impacting 1.4 million devices [7].
"The cost of compliance outweighs the cost of a breach, which risks brand reputation at best and patients' lives at worst." - Heather R. Johnson, Consultant and Writer [1]
Additionally, under Section 524B, incomplete submissions can lead to outright rejection [3]. This new regulatory framework marks a sharp departure from traditional audit-based IT systems, requiring manufacturers to rethink their entire approach to cybersecurity.
By understanding these challenges, manufacturers can take decisive steps to integrate security throughout the entire lifecycle of their devices - from initial design to postmarket management.
Next Steps for Adopting Secure Design Practices
To meet these challenges head-on, manufacturers should embed cybersecurity planning into their development processes from the very start. For example, by integrating a Cybersecurity Management Plan directly into the Software Development Plan, security remains a core focus rather than an afterthought [7].
Some practical steps include:
- Aligning SPDF artifacts with ISO 13485 QMS clauses.
- Maintaining an up-to-date, machine-readable SBOM (e.g., SPDX or CycloneDX) [3][9].
- Establishing a formal postmarket monitoring plan to stay ahead of potential threats [9].
For healthcare delivery organizations juggling multiple vendors and devices, tools like Censinet RiskOps™ can help streamline compliance efforts. These platforms simplify third-party risk assessments, provide cybersecurity benchmarks, and centralize risk management across an entire portfolio of medical devices.
"The rules of the game have changed. The landscape now is to design controls in, period." - Naomi Schwartz, VP of Regulatory Strategy, Medcrypt [1]
FAQs
What counts as a “cyber device” under FDA rules?
A cyber device, according to the Federal Food, Drug, and Cosmetic Act, must satisfy three key conditions:
- It incorporates software, such as firmware or programmable logic, that is authorized by the sponsor.
- It has the capacity to connect to the internet, with the FDA interpreting "internet connectivity" in a broad sense.
- It includes technological features, also authorized by the sponsor, that could be exposed to cybersecurity risks.
What cybersecurity documents does the FDA expect in a premarket submission?
To meet FDA standards, your Quality Management System must incorporate a Secure Product Development Framework (SPDF). This involves submitting specific documentation that demonstrates your commitment to cybersecurity throughout the product lifecycle. Key required artifacts include:
- Software Bill of Materials (SBOM): A detailed inventory of all software components used in your product.
- Postmarket Cybersecurity Management Plan: A strategy for addressing cybersecurity risks after the product is on the market.
- Security Risk Management Report: A comprehensive analysis of identified risks and the measures taken to mitigate them.
In addition, the FDA expects the following to showcase your proactive approach to security:
- Threat models and security architecture documentation.
- Test reports detailing the outcomes of security evaluations.
- Cybersecurity-specific labeling for end-users.
- Evidence of security testing, such as vulnerability assessments and penetration testing results.
These elements work together to demonstrate your ability to identify, address, and reduce potential security risks effectively.
How do we keep an SBOM accurate after the device ships?
To maintain an accurate SBOM after shipment, think of it as a living document. Incorporate SBOM updates directly into your CI/CD pipelines so that every new software release produces an updated, machine-readable inventory. Ensure each SBOM is cryptographically linked to its specific build, creating a dependable audit trail. Additionally, leverage automated tools to scan your device fleet for newly identified CVEs. This approach allows for proactive patching and helps manage risks effectively throughout the entire lifecycle of your devices.
