The FDA's 2025 cybersecurity guidance introduces enforceable rules for medical devices, prioritizing security throughout their lifecycle.
Released on June 27, 2025, this update replaces the 2023 version and implements Section 524B of the FD&C Act. The new standards require manufacturers to meet stricter cybersecurity criteria, including mandatory Software Bills of Materials (SBOMs), continuous vulnerability management, and detailed premarket documentation. Noncompliance can now result in denied market authorization.
Key changes include:
- Expanded scope: Covers devices with internet connectivity, updatable software, and related systems like update servers.
- Lifecycle security: Obligates ongoing threat monitoring, risk-based updates, and customer notifications within 30 days of vulnerability discovery.
- Mandatory SBOMs: Required in premarket submissions to improve transparency and risk management.
- Stricter enforcement: Noncompliance tied directly to market authorization decisions.
This shift aims to strengthen patient safety by enforcing continuous cybersecurity measures while increasing transparency for healthcare organizations.
What Changed in the 2025 Guidance
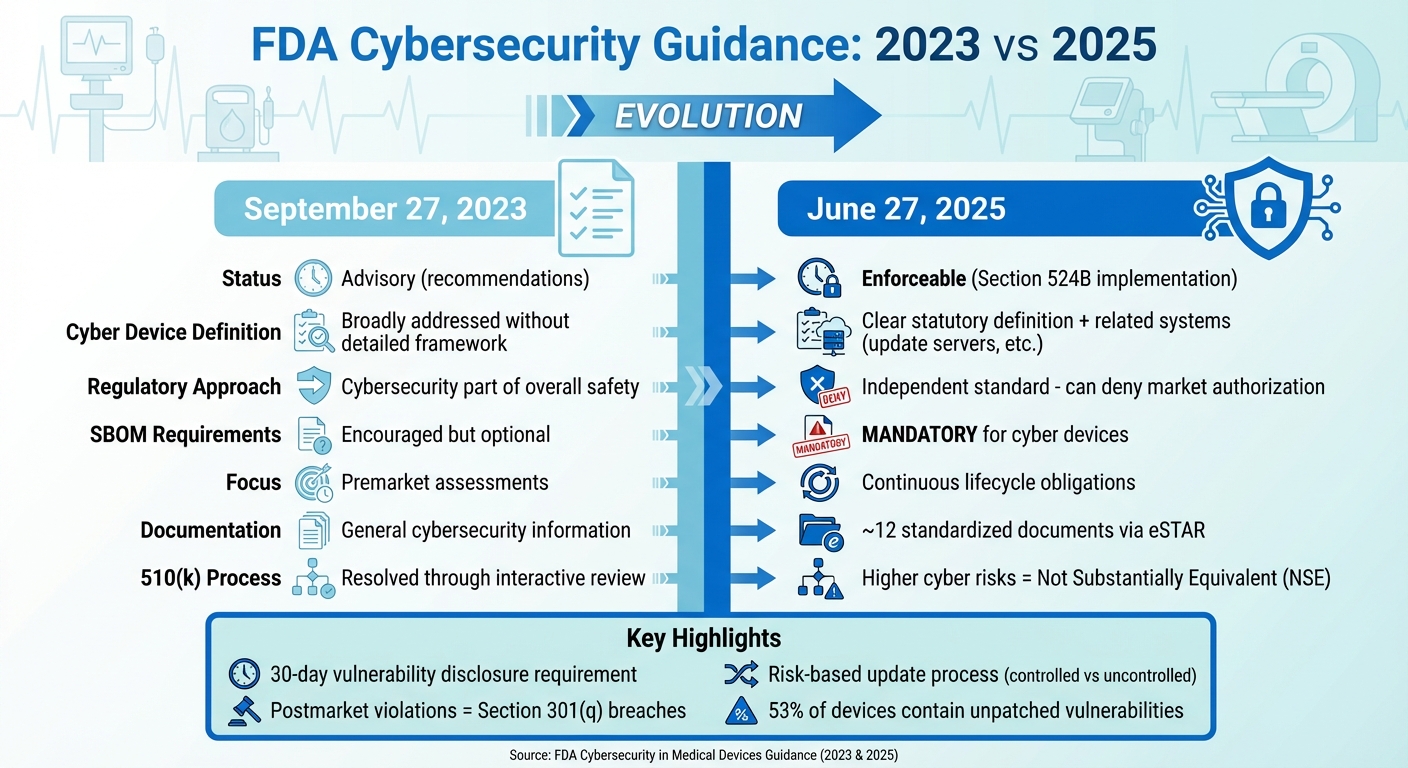
FDA Medical Device Cybersecurity Guidance: 2023 vs 2025 Key Changes
The guidance issued on June 27, 2025, marks a significant shift in how the FDA addresses medical device cybersecurity. Unlike the September 27, 2023 guidance, which treated cybersecurity as advisory, the updated framework introduces enforceable requirements. This change formally incorporates Section 524B of the FD&C Act, establishing "cyber devices" as a distinct category with specific legal obligations. Cybersecurity is no longer just a component of overall safety and effectiveness - it now stands as an independent regulatory standard. This means that a failure to meet cybersecurity requirements could, by itself, result in the denial of market authorization.
One of the most notable updates is the expanded definition of what qualifies as a cyber device. According to the guidance, any device that meets the criteria under Section 524B - whether through internet connectivity, updatable software, or inherent cybersecurity vulnerabilities - is now subject to stricter submission requirements. This broader scope also covers related systems, such as update servers, hospital integration points, connectivity modules, and manufacturer-controlled assets nearing the end of their support period. Examples include devices like infusion pumps with remote monitoring, cloud-connected implantables, robotic surgery systems, and smart ventilators, all of which are now part of this expanded regulatory ecosystem.
Another major change is the shift from a one-time premarket cybersecurity check to an ongoing lifecycle obligation. Manufacturers are now required to maintain active cybersecurity management plans that can adapt to emerging threats. The guidance outlines a risk-based approach to updates, distinguishing between uncontrolled risks that need immediate action and controlled risks that can be addressed during scheduled maintenance windows.
Transparency has also been prioritized in the new guidance. For cyber devices, submitting a Software Bill of Materials (SBOM) is no longer optional - it is now mandatory for premarket submissions. Manufacturers must include detailed architecture, data flow diagrams, and mapped communication protocols, along with a clear disclosure of all communication interfaces, such as USB, Bluetooth, Wi-Fi, cloud APIs, and third-party software integrations. Additionally, the FDA now requires a standardized set of approximately 12 cybersecurity documents to be submitted through the eSTAR process. Noncompliance with these documentation requirements could result in refusal-to-accept or technical screening holds.
| Aspect | 2023 Guidance | 2025 Guidance |
|---|---|---|
| Regulatory Status | Final guidance issued on September 27, 2023, without fully implementing Section 524B. | Final guidance issued on June 27, 2025, explicitly implementing Section 524B and replacing the 2023 version. |
| Cyber Device Definition | Cybersecurity addressed broadly without a detailed statutory framework. | Provides a clear statutory definition linked to Section 524B, including related systems like update servers. |
| Reasonable Assurance | Cybersecurity considered part of overall safety and effectiveness. | Establishes cybersecurity as an independent standard, with deficiencies potentially leading to market denial. |
| SBOM Requirements | SBOMs were encouraged but not required. | SBOMs are now mandatory for cyber devices, impacting premarket submission acceptance. |
| Lifecycle Obligations | Focused on premarket cybersecurity assessments. | Introduces continuous cybersecurity obligations with a risk-based update process. |
| Documentation Set | General cybersecurity information expected. | Requires approximately 12 standardized cybersecurity documents submitted via eSTAR. |
| 510(k) Equivalence | Cybersecurity issues resolved through interactive review. | Devices with higher cyber risks than their predicate may be deemed not substantially equivalent (NSE). |
1. FDA 2023 Cybersecurity Guidance

On September 27, 2023, the FDA released guidance titled "Cybersecurity in Medical Devices: Quality System Considerations and Content of Premarket Submissions." This document established a clear connection between cybersecurity and Quality System Regulation (QSR) design controls, treating cybersecurity as a key element in ensuring the safety and effectiveness of medical devices. Rather than viewing it as just an IT issue, the guidance framed cybersecurity as a critical safety concern. It also set the stage for implementing Section 524B obligations, creating a foundation that influenced the updates introduced in 2025.
Cyber Device Scope
The 2023 guidance, in alignment with Section 524B of the FD&C Act, focused on devices that include software, connect to the Internet, or have features that make them susceptible to cybersecurity threats. This included devices like infusion pumps, implantable cardiac devices, imaging systems, and Software as a Medical Device (SaMD) products. Even devices not explicitly marketed as "connected" were included if their functionality depended on connectivity or software in ways that could affect safety or effectiveness. These definitions established a baseline that was expanded in 2025 to cover additional systems critical to patient safety.
Lifecycle Cybersecurity Assurance
The guidance introduced a secure product development framework to ensure cybersecurity was embedded throughout a device's lifecycle - from initial design to postmarket surveillance. This framework required manufacturers to document their efforts in the Design History File and risk management records. Key elements included secure architecture, threat modeling, secure coding practices, access controls, cryptography, logging mechanisms, and update processes. The expectation was clear: manufacturers needed to show that security was a priority from the very beginning.
Vulnerability Management
Continuous vulnerability management became a critical requirement under this guidance. Manufacturers were tasked with monitoring devices for vulnerabilities, assessing risks, and deploying patches promptly. Vulnerabilities had to be categorized based on their potential to harm patients and the likelihood of exploitation. Risk-based decisions and remediation timelines needed to be documented in quality records and submissions. Additionally, manufacturers were encouraged to maintain open communication with healthcare organizations, providing timely advisories and configuration updates to minimize disruptions to clinical care.
Transparency and SBOM Requirements
To promote transparency, the FDA encouraged manufacturers to include a Software Bill of Materials (SBOM) in their premarket submissions. Many healthcare providers are now leveraging SBOM data through platforms like Censinet RiskOps™ to assess third-party risks. The guidance also emphasized the importance of providing clear labeling and technical documentation to outline security controls, supported configurations, update mechanisms, and any known limitations. These transparency measures laid the groundwork for stricter SBOM requirements in subsequent updates, signaling the FDA's shift from offering recommendations to enforcing cybersecurity mandates.
2. FDA 2025 Cybersecurity Guidance
In June 2025, the FDA issued its final guidance titled "Cybersecurity in Medical Devices: Quality System Considerations and Content of Premarket Submissions," replacing the 2023 version [6][7][8]. This update shifts cybersecurity from being a set of voluntary recommendations to enforceable legal requirements under Section 524B of the FD&C Act. It places cybersecurity as an ongoing responsibility, requiring manufacturers to actively manage plans that address evolving threats. Failure to maintain cybersecurity measures postmarket can now result in violations under Section 301(q), which classifies such lapses as prohibited acts [1][4]. Below, we’ll break down the key changes and their practical implications.
Cyber Device Scope
The updated guidance expands the definition of what qualifies as a "cyber device" under Section 524B. It applies to devices that include software, connect to the internet (directly or indirectly), or have technological features that could be exploited, potentially harming patients [1][4][6]. Examples include networked imaging systems and wearable ECG monitors linked to mobile apps [1][2].
In addition, the guidance broadens compliance requirements to include servers, cloud infrastructure, and network integration points, recognizing that vulnerabilities in these systems can directly impact patient safety [1][5].
Lifecycle Cybersecurity Assurance
The FDA now requires manufacturers to maintain continuous cybersecurity assurance throughout a device’s lifecycle. This involves implementing a Secure Product Development Framework (SPDF) across design, validation, and risk management stages [1][2][6]. Premarket submissions for cyber devices must include a detailed cybersecurity management plan, along with about 12 supporting documents submitted via eSTAR. These documents cover:
- SPDF descriptions
- Threat models and risk assessments
- Security architecture and data-flow diagrams
- Access control and cryptography details
- Security testing plans and results
- Update and patching strategies
- Vulnerability monitoring procedures
- Labeling with security configuration instructions
Failure to provide these elements can lead to Refuse to Accept (RTA) decisions or technical screening holds, delaying approval for U.S. market entry [2][6]. Postmarket, manufacturers are expected to ensure continuous monitoring, disclose vulnerabilities, provide timely updates, and document processes that meet quality system requirements [2][3][4].
Vulnerability Management
The 2025 guidance reinforces a risk-based approach to managing vulnerabilities. Manufacturers must classify vulnerabilities as either uncontrolled (requiring immediate action) or controlled (addressed during scheduled maintenance), depending on their impact on safety and essential performance [1].
Manufacturers are advised to notify customers of vulnerabilities within about 30 days of discovery and provide appropriate fixes or patches based on the risk level [1]. With 53% of devices reportedly containing unpatched vulnerabilities [1], the guidance outlines a comprehensive process that includes:
- Accepting vulnerability reports (including those from external researchers)
- Conducting technical assessments and risk scoring
- Prioritizing and testing fixes
- Delivering secure updates
- Communicating with customers
These steps must integrate with quality systems and complaint/CAPA procedures where applicable [1][4][7]. The guidance also stresses the importance of defense-in-depth strategies, such as secure default configurations, least-privilege access, multi-factor authentication (MFA), strong encryption aligned with NIST and FIPS 140-3, segmentation of critical functions, and telemetry to detect anomalies [1][3][7].
Transparency and SBOM Requirements
Transparency is now a cornerstone of the FDA’s 2025 guidance, with a focus on software bill of materials (SBOM) requirements for cyber devices under Section 524B [2][6][8]. Manufacturers must submit detailed SBOMs that include both first- and third-party components, enabling quick assessments of exposure to vulnerabilities [2][7][8].
SBOMs should be treated as living documents, updated with every software release, and formatted in standard structures like SPDX or CycloneDX to support security teams and healthcare organizations [2][8]. The guidance also emphasizes clear communication of known vulnerabilities, security features, supported configurations, and update practices in device labeling. This transparency helps healthcare providers integrate devices into their own risk management and patching workflows [1][2][6].
Platforms like Censinet RiskOps™ are already helping U.S. healthcare providers use SBOM data to map vulnerabilities to enterprise risk registers, making it easier for vendors and customers to collaborate on fixes and communication.
sbb-itb-535baee
Advantages and Challenges
The transition from the 2023 to the 2025 guidance brings notable advancements in security while raising the bar for compliance among manufacturers and healthcare organizations. Unlike the 2023 guidance, which primarily served as recommendations, the 2025 version enforces security requirements under Section 524B. This change makes cybersecurity compliance a condition for market authorization, redefining the criteria for device approval. While this shift strengthens patient protections and provides clearer expectations, it also introduces operational challenges. Let’s break down the key advantages and hurdles.
The 2025 guidance gives manufacturers a clearer framework for meeting cybersecurity expectations. By aligning with IMDRF Principles, it supports global consistency, making international compliance more straightforward. For healthcare delivery organizations, the new requirements - such as Software Bills of Materials (SBOMs), timelines for vulnerability disclosures, and detailed cybersecurity management plans - enhance transparency. This richer data supports more effective third-party risk assessments and enables tools like Censinet RiskOps™ to automate vendor benchmarking and risk mapping across clinical settings.
But with these benefits come challenges. Manufacturers now face a significant compliance workload, including a 12-document eSTAR submission requirement that spans everything from threat modeling to vulnerability communication. Missing any of these documents could result in Refuse to Accept (RTA) decisions or technical screening holds, delaying market entry in the U.S. Stricter requirements may also force late-stage design changes, which can disrupt time-to-market. Smaller manufacturers, or those relying on outdated quality management systems, often struggle to implement Secure Product Development Frameworks (SPDFs) and maintain accurate SBOMs across intricate supply chains.
Here’s a comparison of how the 2023 and 2025 guidances address vulnerability management and transparency:
| Aspect | 2023 Guidance | 2025 Guidance |
|---|---|---|
| Vulnerability Management | Encouraged disclosure and patching but lacked specific timelines or communication standards. | Recommends a 30-day disclosure for significant vulnerabilities and differentiates between controlled and uncontrolled risks. |
| Transparency (SBOM & Documentation) | Recommended SBOMs and threat modeling but didn’t tie them tightly to regulatory decisions. | Requires SBOMs in premarket submissions for cyber devices and mandates 12 cybersecurity documents, with omissions leading to RTA. |
| Regulatory Enforcement | Provided expectations but didn’t explicitly link them to authorization denials. | Links cybersecurity compliance to Section 524B, allowing devices to be deemed "not substantially equivalent" (NSE) due to cyber risks. |
| Implementation Burden | Flexible but open to interpretation, with variability in industry adoption. | Sets a higher compliance standard, demanding continuous cybersecurity management, formal SPDFs, and sustained resource investments. |
For healthcare organizations, the 2025 guidance adds complexity to cybersecurity data management. Security teams must now map SBOM components to known vulnerabilities, align vendor risk classifications with internal clinical impact assessments, and maintain thorough records for regulatory and insurance purposes. While these measures enhance patient safety, they also require advanced tools and automation to handle the increased workload. Platforms designed for risk management can simplify these tasks, but both manufacturers and healthcare providers will need to adjust their practices to keep pace with these evolving regulations.
Conclusion
The FDA's transition from the 2023 to the 2025 cybersecurity guidance represents a major shift in how medical device security is regulated in the United States. While the 2023 guidance introduced foundational cybersecurity principles and lifecycle concepts, the 2025 guidance elevates these ideas into enforceable requirements under Section 524B of the FD&C Act. Cybersecurity is no longer just a factor in ensuring safety and effectiveness - it is now a standalone regulatory requirement that spans the entire lifecycle of a device, from its design to decommissioning. Compliance is now mandatory, with postmarket violations classified as regulatory breaches under Section 301(q). This shift paves the way for a unified approach to managing risks across design, deployment, and clinical use.
Manufacturers must now prioritize cybersecurity as an integral part of their quality systems. This involves adopting a Secure Product Development Framework (SPDF) that includes threat modeling, secure coding practices, and continuous vulnerability monitoring. A living Software Bill of Materials (SBOM) must be maintained, along with comprehensive cybersecurity documentation submitted via eSTAR to avoid Refuse to Accept decisions. Ongoing cybersecurity assurance, robust vulnerability disclosure processes, and an updated cybersecurity management plan are now essential for compliance.
Healthcare delivery organizations also need to adapt to these changes. Procurement processes should now require SBOMs, patch service-level agreements (SLAs), and strong vulnerability disclosure protocols. Incorporating device cybersecurity into broader enterprise risk management strategies - such as managing asset inventories, implementing network segmentation, and coordinating patching efforts - will be key to ensuring patient safety. With enhanced data from manufacturers, third-party risk assessments and clinical decision-making can become more streamlined. Tools like Censinet RiskOps™ can assist in automating vendor evaluations and mapping risks across clinical environments, simplifying the management of these new requirements.
Both manufacturers and healthcare organizations should perform gap assessments to align their current practices with the 2025 guidance and Section 524B standards. Strengthening cybersecurity management plans, implementing technical controls like multi-factor authentication and strong cryptographic measures, and eliminating vulnerabilities like hardcoded passwords will be critical. Establishing governance structures that bring together clinical, IT, security, and regulatory teams can help avoid delays in device clearance and reduce enforcement risks. The 2025 guidance is more than a compliance mandate - it’s an opportunity to create a culture of continuous risk management that protects patients while keeping pace with the ever-evolving threat landscape.
FAQs
What are the main updates in the FDA's 2025 cybersecurity guidance compared to 2023?
The FDA’s 2025 cybersecurity guidance introduces some important changes, emphasizing preventing risks, real-time threat management, and stricter standards for medical device cybersecurity. Unlike the 2023 guidelines, this update places a stronger focus on emerging technologies like AI and automation.
Here’s what’s different:
- A stronger focus on preventing cybersecurity risks rather than just responding to incidents after they occur.
- Clearer guidelines outlining manufacturers' responsibilities for managing cybersecurity risks after devices are on the market.
- Improved protocols to handle threats as they happen, ensuring quicker responses to potential vulnerabilities.
These updates reflect the increasing complexity of cybersecurity in healthcare, aiming to safeguard both patient safety and sensitive data more effectively.
What changes in the 2025 FDA cybersecurity guidance affect medical device approvals?
The 2025 FDA cybersecurity guidance brings in tougher standards for managing risks and securing medical devices. These changes aim to prioritize patient safety and safeguard sensitive data, pushing manufacturers to incorporate strong cybersecurity measures right from the design phase.
Meeting these updated guidelines could streamline the approval process for manufacturers. Devices that show compliance and preparedness might not only move through regulatory hurdles faster but also foster stronger trust with regulators and healthcare providers - potentially speeding up their market launch.
What does the 2025 FDA guidance require for a Software Bill of Materials (SBOM)?
The FDA's 2025 cybersecurity guidance places a strong emphasis on the Software Bill of Materials (SBOM), requiring it to be thorough, precise, and regularly updated. This document must list every software component and dependency within medical devices, including third-party and open-source software.
The goal here is to boost visibility and reinforce risk management. By detailing these components, manufacturers can more easily identify vulnerabilities and enhance supply chain security. This approach also supports improved cybersecurity measures and ensures compliance with regulatory expectations.
